Introduction
Malignant giant cell tumor of bone (GCTB) is a
clinicopathologically defined diagnostic concept characterized by
the presence of multinucleated giant cells and an aggressive
clinical behavior associated with a high risk of metastasis or
local recurrence (1). Malignant GCTB
is treated by wide resection; however, the prognosis is unfavorable
(2).
H3 histone family member 3A (H3F3A) encodes
for a H3.3 protein. GCTB is genetically characterized by a highly
recurrent mutation in H3F3A, with the G34W mutation being
the most common (1-3).
The H3.3 G34W mutation is highly specific for GCTB, and almost all
histological mimics lack this genetic signature (4,5). The
loss of H3.3K36me3 on mutant H 3.3 alters the deposition of the
repressive H3K27me3 mark from intergenic to genic regions, beyond
areas of H3.3 deposition. This alteration promotes the
redistribution of other chromatin marks and aberrant transcription,
altering cell fate in mesenchymal progenitors and hindering
differentiation (6). Previous
studies have reported that the H3F3A mutations can also be
detected in malignant GCTB (5,7).
However, some malignant GCTBs have been found to be negative for
H3F3A mutations, even though the paired GCTB component has
been found positive for H3F3A mutations (5). Other reports suggested that TP53
mutation, KRAS/HRAS mutation, TERT mutation,
KDM4B/KDM6A loss, or H3K27me3 loss may be associated with
the malignant progression of GCTB (8-11).
However, oncogenic events in H3F3A wild-type malignant GCTB
remain unknown.
In the present study, it was hypothesized that
as-yet-unknown molecular events participate in the progression of
malignant GCTB. Therefore, the present study analyzed genomic
alterations in 8 cases of clinicopathologically diagnosed malignant
GCTB using the Center for Cancer Genomics and Advanced Therapeutics
(C-CAT) genomic database.
Patients and methods
Study design
The present study retrospectively analyzed the
results of genomic profiling tests using extracted data from a
Japanese nationwide genomic database (C-CAT).
Comprehensive genomic profiling and
the C-CAT database
In Japan, insurance coverage for the cancer
comprehensive genomic profiling (CGP) test was implemented in June,
2019 (12,13). In total, three types of CGP tests are
available through the national health insurance system for patients
with advanced solid tumors who have completed standard chemotherapy
or for whom no appropriate standard chemotherapy is available: The
Foundation One® CDx (F1CDx; Foundation Medicine, Inc.)
test, Foundation One® Liquid CDx (F1LCDx; Foundation
Medicine, Inc.) test and the OncoGuide NCC Oncopanel System
(https://www.ncc.go.jp/en/information/press_release/20190717/20190717152024.html).
C-CAT information is available elsewhere (13). Briefly, C-CAT was established at the
National Cancer Center as an organization that collects and
facilitates the use of data derived from CGP tests (12,13).
C-CAT collects CGP results and clinical information for almost all
patients undergoing CGP after obtaining written informed consent.
These data can be used in clinical trials and drug development
following approval by both the institutional review board and
C-CAT. As of March, 2023, >50,000 patients with advanced-stage
cancer have undergone CGP tests since June, 2019.
Data extraction
A search was made on the anonymized C-CAT database
of genomic and clinical information on patients with malignant bone
tumors. The clinical data in C-CAT include age, sex, histology,
treatment before and after CGP tests, drug response and type of CGP
test used. A total of 384 samples of genomic data were detected in
the malignant bone tumor cohort of C-CAT from 2019 to 2022. Of
these, eight malignant GCTB datasets were extracted for the present
study. In other words, the genomic data of sequencing analysis
results were already available and actual sequencing or mutation
analysis was not performed during the present study. All eight
samples were sequenced by F1CDx. Information on gene alterations
was annotated using Cancer Knowledge Databases, such as OncoKB,
ClinVar and COSMIC, etc, at C-CAT (13).
The F1CDx assay employs formalin-fixed
paraffin-embedded tumor tissue samples obtained via biopsy or
surgical procedure, with pathologists selecting suitable tumor
specimens for testing (details available at https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx).
All histological diagnoses were made using morphology,
immunohistochemistry and molecular data by specialized clinicians
and pathologists in each hospital. The present study was approved
by the Institutional Review Board of the University of Tokyo
(Tokyo, Japan; approval no. 2021341G) and the C-CAT information
utilization review committee (proposal control no. CDU2022-026
N).
Statistical analysis
A Student's t-test test was used to compare the
quantitative variables between two groups. A two-tailed probability
(P)-value <0.05 was considered to indicate a statistically
significant difference. Statistical analyses were performed using
SPSS version 22.0 software (IBM Corp.).
Results
Clinical characteristics
The clinical characteristics of the 8 patients with
malignant GCTB whose data were analyzed in the present study are
summarized in Table I. The median
age of the patients was 33 years, and 5 patients (63%) were male. A
total of seven samples were collected from the primary sites, and
one sample was collected from a metastatic lesion. Of the 8
patients included, 5 (63%) patients had metastasis, including to
the lung, bone, peritoneum, spinal cord, soft tissue, or adrenal
gland, when the F1CDx test was performed. A total of 5 patients
received chemotherapy (cisplatin, doxorubicin, or ifosfamide) or
denosumab. At the time of the final follow-up data, 3 patients had
succumbed to the disease.
 | Table IClinical and genomic characteristics
of the patient whose data were analyzed in the present study. |
Table I
Clinical and genomic characteristics
of the patient whose data were analyzed in the present study.
| Case no. | Sex | Age, years | H3F3A
mutation | Metastasis | Drug | Outcome |
|---|
| 1 | M | 35 | Mutant | NA | NA | NA |
| 2 | F | 25 | Mutant | Lung, spinal cord,
soft-tissue, adrenal grand | CDDP, DOX | NA |
| 3 | M | 48 | Mutant | Lung | CDDP, DOX | NA |
| 4 | F | 30 | Mutant | Lung | CDDP, DOX | DOD |
| 5 | F | 9 | Wild | Peritoneum | No | NA |
| 6 | M | 7 | Wild | No | IFO | Alive |
| 7 | M | 73 | Wild | Bone | Denosumab | DOD |
| 8 | M | 41 | Wild | No | NA | DOD |
Comprehensive genomic profiling
test
A total of 78 mutations were detected (data not
shown). Among these, 26 mutations were annotated as likely or known
oncogenic alterations, with an average of 3.1 (26 of 8) alterations
per sample (Table II). The
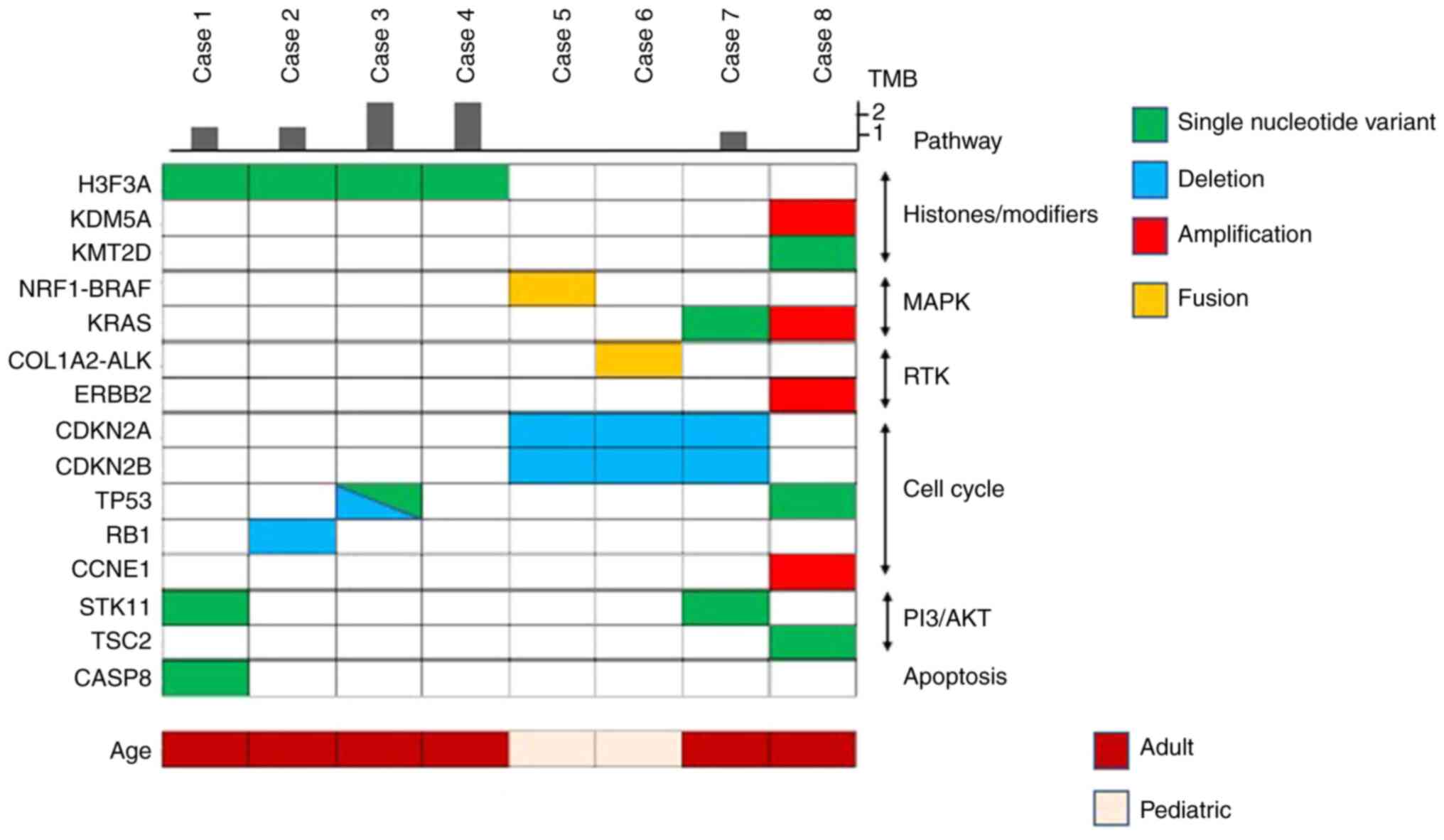
oncoprint is depicted in Fig. 1.
Single-nucleotide variants accounted for 46% (12 of 26) of the
alterations, and copy number alterations (deletion and
amplification) and rearrangements (fusion) accounted for 46% (12 of
26) and 8% (2 of 26), respectively. H3F3A G34W mutations
(hg38, chr1: 226064454 G>T) and G34L mutation (hg38, chr1:
226064454 GG>CT) were found in 3 patients and 1 patient,
respectively. In 50% of the cases with H3F3A mutation, other
co-occurring mutations were related to cell cycle regulators
(TP53 or RB1). mTOR pathway gene alterations
(STK11 or TSC2) were detected in 3 of the 8 (38%)
cases (Fig. 1 and Table II).
| Table IIOncogenic alterations identified in
the present study. |
Table II
Oncogenic alterations identified in
the present study.
| Case | Gene | Chromosome | Genomic
locations | Reference | Base change | Amino acid
change | Mutation allele
frequency | TMB (Muts/Mb) | MSI |
|---|
| Case 1 | CASP8 | 2 | 201266689 | G | A | R68Q | 0.53 | 1.26 | Stable |
| | H3F3A | 1 | 226064454 | G | T | G34W | 0.08 | | |
| | STK11 | 19 | 1223126 | C | G | F354L | 0.54 | | |
| Case 2 | RB1 | 13 |
48411294-48515183 | - | Deletion | | | 1.26 | Stable |
| | H3F3A | 1 | 226064454 | GG | CT | G34L | 0.41 | | |
| Case 3 | TP53 | 17 |
7673177-7703534 | - | Deletion | | | 2.52 | |
| | H3F3A | 1 | 226064454 | G | T | G34W | 0.20 | | |
| | TP53 | 17 | 7674241 | G | A | S241F | 0.07 | | |
| Case 4 | H3F3A | 1 | 226064454 | G | T | G34W | 0.14 | 2.52 | Stable |
| Case 5 | CDKN2A | 9 |
21968170-21994454 | - | Deletion | | | 0 | Stable |
| | CDKN2B | 9 |
22002171-22010785 | - | Deletion | | | | |
| | NRF1-BRAF | 7:7 |
140789425:129699940 | - | Fusion | | | | |
| Case 6 | CDKN2A | 9 |
21968170-21994454 | - | Deletion | | | 0 | Stable |
| | CDKN2B | 9 |
22002171-22010785 | - | Deletion | | | | |
| | COL1A2-ALK | 2:7 |
29227044:94417378 | - | Fusion | | | | |
| Case 7 | CDKN2A | 9 |
21954945-21998003 | - | Deletion | | | 1 | Stable |
| | CDKN2B | 9 |
21998749-22069275 | - | Deletion | | | | |
| | KRAS | 12 | 25245350 | C | T | G12D | 0.47 | | |
| | STK11 | 19 | 1223126 | C | G | F354L | 0.48 | | |
| Case 8 | CCNE1 | 19 |
29763011-29869731 | - | Amplification | | | 0 | Stable |
| | ERBB2 | 17 |
39651436-39777579 | - | Amplification | | | | |
| | KDM5A | 12 | 285455-389091 | - | Amplification | | | | |
| | KRAS | 12 |
25191796-25295283 | - | Amplification | | | | |
| | KMT2D | 12 | 49050247 | TC | T | D1114fs*5 | 0.05 | | |
| | TP53 | 17 | 7674903 | TTC | T | R209fs*6 | 0.43 | | |
| | TSC2 | 16 | 2086815 | TTT | T | F1645fs*7 | 0.14 | | |
In 75% of the cases without H3F3A mutation
(case nos. 5, 7 and 8; Table II),
mitogen-activated protein kinase (MAPK) signaling pathway gene
alterations were found (KRAS single nucleotide variant, KRAS
amplification, nuclear respiratory factor 1
(NRF1)-BRAF fusion). Moreover, the collagen type I
alpha 2 chain (COL1A2)-ALK fusion was detected in the
remaining one case (case no. 6). All 4 cases without H3F3A
mutation (case nos. 5-8) had gene alterations related to cell cycle
regulators [cyclin-dependent kinase inhibitor 2 (CDKN2)A and
CDKN2B loss, TP53 mutation and cyclin E1
(CCNE1) amplification]. OF note, 1 case had alterations in
epigenetic modulator genes, such as KDM5A or KMT2D
(Fig. 1 and Table II).
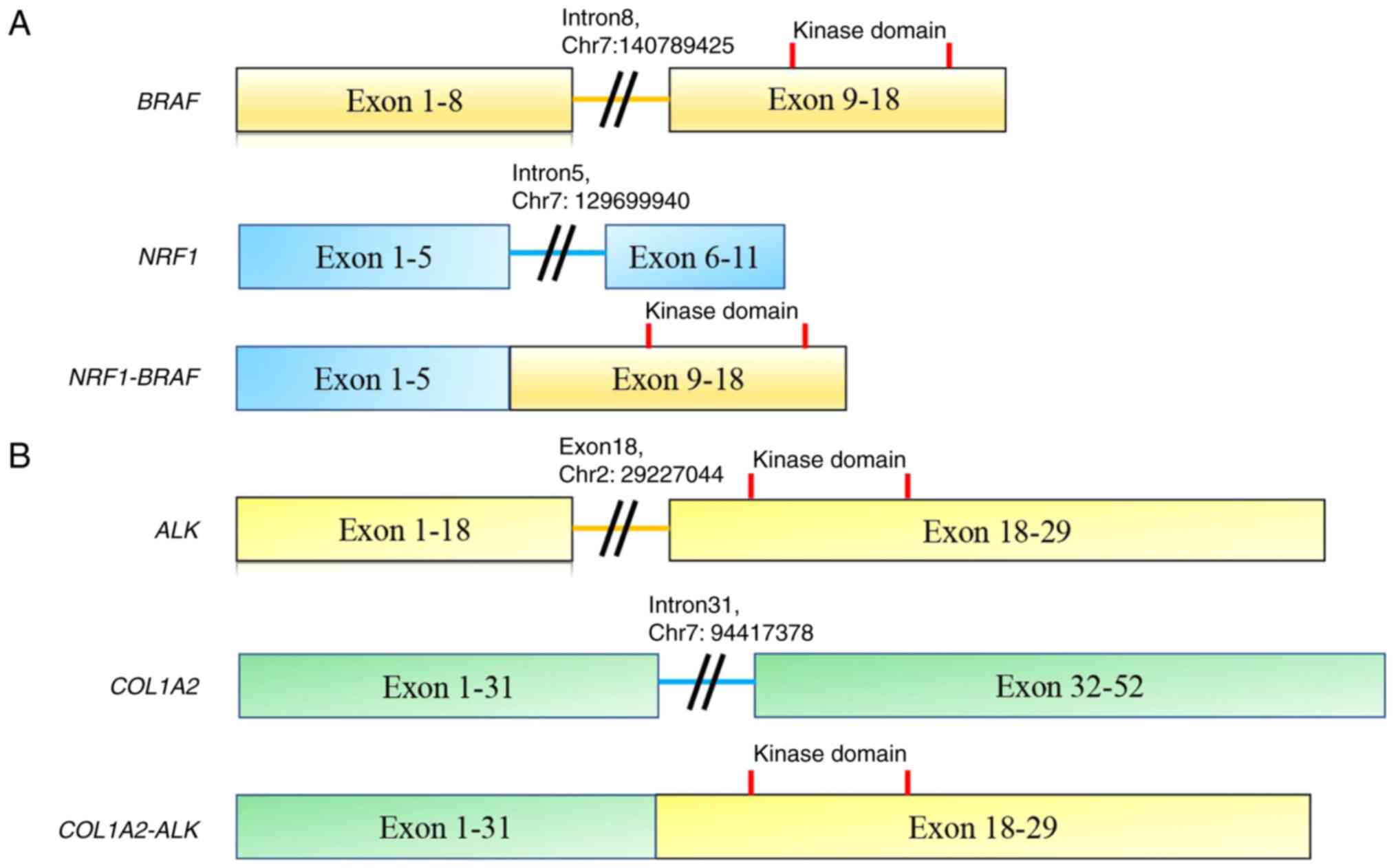
NRF1 intron 5 (chr7: 129699940) was fused
with BRAF intron 8 (chr7: 140789425) (Fig. 2). The COL1A2-ALK rearrangement
comprised intron 31 of COL1A2 (chr7: 94417378) and exon 18
of ALK (chr2: 29227044). The kinase domains of both
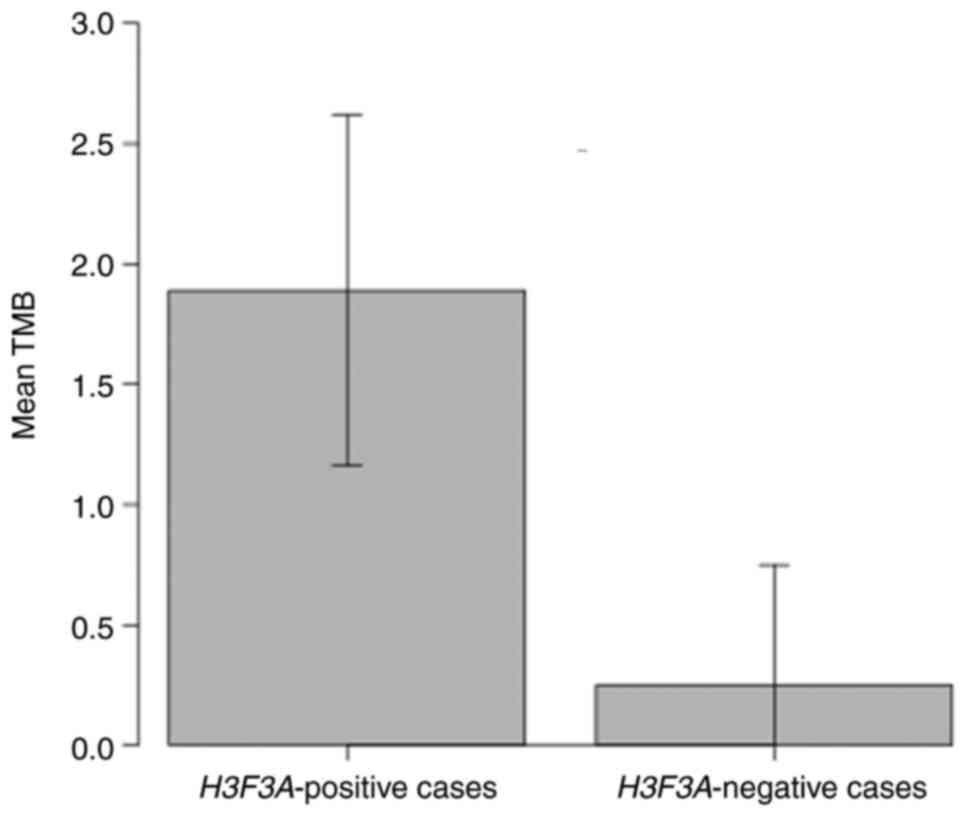
predicted proteins were retained. The tumor mutation burden (TMB)
was significantly lower in the samples without H3F3A
mutation (case 5, 6, 7, 8) than in the samples with H3F3A
mutation (case 1, 2, 3, 4) (Student's t-test, mean 0.25 vs. mean
1.89, P=0.01, Fig. 3). Of the 8
cases analyzed herein, the patients with kinase fusion had unique
characteristics, such as a younger age (9 and 7 years) and a lower
TMB (both, 0 muts/Mb) compared to the fusion-negative cases. No
patients were enrolled in a trial or off-label use of an approved
drug due to trial ineligibility, poor performance status, or
unknown reasons.
Discussion
Using a large genomic database (C-CAT database), the
present study analyzed the genomic alterations of
clinicopathologically diagnosed malignant GCTB. A total of 4 cases
had H3F3A mutations and MAPK signaling pathway gene
alterations were found in 75% of the cases without H3F3A
mutation. The most frequent concurrent gene alterations were
related to cell cycle regulators, including TP53,
RB1, CDKN2A/B and CCNE1 (75%, 6 of 8 cases).
Potentially targetable fusion genes (NRF1-BRAF and
COL1A2-ALK) were also detected.
Malignant GCTB is difficult to characterize due to
its rarity, broad histological spectrum and the occasional presence
of abundant giant cells in unrelated sarcomas (5). H3F3A mutations are detected in
benign and malignant GCTB. Although a few H3F3A
mutation-negative malignant GCTBs have been reported, none have
been thoroughly investigated (5).
Herein, MAPK signaling pathway alterations were observed in
patients with H3F3A wild-type tumors. Consistent with these
findings, KRAS G12V was previously detected in malignant
GCTB (8). HRAS mutations were
also previously found in two cases of malignant GCTB (9), indicating the importance of RAS family
mutations in the malignant progression of GCTB. KRAS is a
frequently mutated oncogene in numerous types of cancer, including
non-small cell lung cancer, colorectal cancer and pancreatic ductal
adenocarcinoma (14-16).
KRAS mutations cause conformational changes in
KRAS-binding Raf proteins, activating downstream effectors
involved in cellular growth, differentiation and survival (17).
Cell cycle regulator gene alterations were
frequently found in the cohort in the present study. A previous
study reported that 80% (4 of 5 cases) of pleomorphic or
epithelioid cell-predominant malignant GCTB were positive for TP53
nuclear accumulation (11). Fittall
et al (10) identified driver
events in malignant bone tumors with H3F3A mutation using
comprehensive genomic and methylation profiling. Malignant
progression necessitated additional genetic mutations, such as
TP53 mutations, which was consistent with the findings of
the present study. In contrast to the findings of the present
study, Fittall et al (10)
also detected recurrent TERT promoter mutation.
The single nucleotide alteration of H3F3A
induces epigenomic alterations with implications for the
development of stromal cells and the tumorigenic process in benign
GCTB (18). H3F3A mutations
are plausibly crucial oncogenic event in malignant GCTB. Other
histone modifier gene alterations, such as KDM5A or
KMT2D were detected in the present study, although further
studies are required to confirm the importance of these
alterations. Biallelic losses of histone lysine demethylase,
KDM4B or KDM5A were previously also found (10). Ishihara et al (11) reported that 3 of 4 (75%) cases of
spindle cell-predominant malignant GCTBs were negative for H3K27me3
and EZH2 mutation was found in 1 case, which suggested that
the dysfunction of histone methylation, as evidenced by the loss of
H3K27me3, may play a key role in the malignant progression of GCTB
(11). In contrast to these
findings, the EZH2 mutation was not detected in the present
study. The role of the loss of H3K27me3 in malignant GCTB warrants
further investigation.
Two fusion genes (NRF1-BRAF and
COL1A2-ALK) need to be carefully validated following the
pathology rereview. BRAF or ALK fusion has not yet
been reported in malignant GCTB. The NRF1-BRAF fusion gene
was previously detected in 2 cases of anaplastic pleomorphic
xanthoastrocytoma (PXA) and urothelial carcinoma (19,20). In
the case of PXA, the predicted fusion protein contained exons 1-5
of NRF1 and the serine/threonine kinase domain of
BRAF. Immunohistochemistry confirmed the robust activation
of the MAPK signaling pathway. The loss of CDKN2A was also
found in the tumor (19). Another
case involved a high-grade papillary urothelial carcinoma in the
renal pelvis that had invaded the renal parenchyma and spread to
the lymph nodes, liver, cervical and lumbar spine and humerus.
F1CDx examined a biopsy of the liver lesion and discovered the
NRF1-BRAF fusion. On the basis of the genomic results, the
patient opted to begin a trial of trametinib (Mekinist), a
second-generation MEK inhibitor. Following 2.5 months of treatment,
an MRI scan revealed that the tumor had shrunk by 48.4% (20). In the present study, in case 5,
NRF1 intron 5 (chr7: 129699940) and BRAF intron 8
(chr7: 140789425) were involved, retaining the serine/threonine
kinase domain of BRAF. Although the confirmation of the
fusion transcript and immunohistochemistry for MAPK signaling
pathway activation is desirable, the case in the present study may
be a candidate for targeted therapy, including MEK and/or
BRAF inhibitors.
The COL1A2-ALK fusion has been found in
ALK-positive histiocytosis (21). Chang et al (21) reported 10 patients with
ALK-positive histiocytosis, 6 of whom had disseminated
disease: A total of 5 cases developed in early infancy with
eventual disease resolution, and the 6th patient presented at 2
years of age and succumbed due to intestinal, bone marrow and brain
involvement (21). The other 4
patients had localized disease involving the nasal skin, foot,
breast and intracranial cavernous sinus; the first 3 patients had
no recurrence following surgical resection, and the cavernous sinus
lesion resolved completely with the ALK inhibitor,
crizotinib (21). The association
between case 6 in the present study and ALK-positive
histiocytosis is unknown as the pathology was not rereviewed.
Touton-type giant cells have been found in ALK-positive
histiocytosis (22), which could
lead to a misdiagnosis of malignant GCTB. The findings presented
herein suggest that potentially targetable ALK fusions are
present in a subset of cases clinicopathologically diagnosed with
malignant GCTB.
The present study has several limitations which
should be mentioned. First, the pathology was not rereviewed by a
sarcoma pathologist, which may have resulted in some
misclassifications. Malignant GCTB in young patients is rare. In
particular, two fusion genes should be carefully validated after
the pathology re-review by sarcoma pathologists. These two fusion
genes may be detected in the resembling tumors, which contain giant
cells, apart from malignant giant cell tumor. Second, the C-CAT
database lacked the details of fusion gene (in-frame or out-frame).
Third, data on whether the tumors were primary or secondary
malignant GCTB were not available, and mutation patterns in primary
and secondary tumors may differ. However, the real-world data used
provide a unique perspective on genomic alterations in
clinicopathologically diagnosed malignant GCTB. Fourth, the lack of
matched normal control DNA may result in the inclusion of germline
mutations inadvertently.
In conclusion, the findings of the present study
suggest that MAPK pathway alterations are crucial in
H3F3A-wild type malignant GCTB. The most frequent oncogenic
event was gene alterations related to cell cycle regulators.
Potentially targetable BRAF or ALK fusion may be
detected in a subset of cases clinicopathologically diagnosed with
malignant GCTB that lack H3F3A mutation; however, the
careful validation of two fusion genes and a pathology review need
to be performed. The real-world findings highlight a unique
perspective on genomic alterations in clinicopathologically
diagnosed malignant GCTB.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
YT and HKo collected and analyzed the data. YT, HKa,
ASU, KOd, HKo, and ST wrote the manuscript. All authors examined
and edited the manuscript. YT, LZ, TH, YI, HKa, ASU, KOd, KOk, HKo
and ST were involved in the conception and design of the study. All
authors have read and approved the final manuscript. YT and HKo
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the University Tokyo (Tokyo, Japan; approval no.
2021341G) and the C-CAT information utilization review committee
(proposal control no. CDU2022-026 N). Patient consent was waived
due to the retrospective nature of the study and as the analysis
used anonymous clinical data.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Flanagan AM, Larousserie F, O'Doneell PG
and Yoshida A: Giant Cell Tumor of Bone. In: WHO Classification of
Tumours Editorial Board. Soft tissue and bone tumours. 5th edition.
International Agency for Research on Cancer, Lyon, 2020.
|
|
2
|
Bertoni F, Bacchini P and Staals EL:
Malignancy in giant cell tumor of bone. Cancer. 97:2520–2529.
2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Behjati S, Tarpey PS, Presneau N, Scheipl
S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et
al: Distinct H3F3A and H3F3B driver mutations define
chondroblastoma and giant cell tumor of bone. Nat Genet.
45:1479–1482. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Cleven AH, Hocker S, Briaire-de Bruijn I,
Szuhai K, Cleton-Jansen AM and Bovee JV: Mutation analysis of H3F3A
and H3F3B as a diagnostic tool for giant cell tumor of bone and
chondroblastoma. Am J Surg Pathol. 39:1576–1583. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yoshida KI, Nakano Y, Honda-Kitahara M,
Wakai S, Motoi T, Ogura K, Sano N, Shibata T, Okuma T, Iwata S, et
al: Absence of H3F3A mutation in a subset of malignant giant cell
tumor of bone. Mod Pathol. 32:1751–1761. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Khazaei S, Jay ND, Deshmukh S, Hendrikse
LD, Jawhar W, Chen CCL, Mikael LG, Faury D, Marchione DM, Lanoix J,
et al: H3.3 G34W promotes growth and impedes differentiation of
Osteoblast-Like mesenchymal progenitors in giant cell tumor of
bone. Cancer Discov. 10:1968–1987. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Amary F, Berisha F, Ye H, Gupta M,
Gutteridge A, Baumhoer D, Gibbons R, Tirabosco R, O'Donnell P and
Flanagan AM: H3F3A (Histone 3.3) G34W Immunohistochemistry: A
reliable marker defining benign and malignant giant cell tumor of
bone. Am J Surg Pathol. 41:1059–1068. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Donigian S, Whiteway SL, Hipp SJ, Lybeck D
and Clark RO: Malignant giant cell tumor of bone with a KRAS G12V
mutation. J Pediatr Hematol Oncol. 44:e268–e271. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Oda Y, Sakamoto A, Saito T, Matsuda S,
Tanaka K, Iwamoto Y and Tsuneyoshi M: Secondary malignant
giant-cell tumour of bone: Molecular abnormalities of p53 and H-ras
gene correlated with malignant transformation. Histopathology.
39:629–637. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fittall MW, Lyskjaer I, Ellery P, Lombard
P, Ijaz J, Strobl AC, Oukrif D, Tarabichi M, Sill M, Koelsche C, et
al: Drivers underpinning the malignant transformation of giant cell
tumour of bone. J Pathol. 252:433–440. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ishihara S, Yamamoto H, Iwasaki T, Toda Y,
Yamamoto T, Yoshimoto M, Ito Y, Susuki Y, Kawaguchi K, Kinoshita I,
et al: Histological and immunohistochemical features and genetic
alterations in the malignant progression of giant cell tumor of
bone: A possible association with TP53 mutation and loss of H3K27
trimethylation. Mod Pathol. 35:640–648. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mukai Y and Ueno H: Establishment and
implementation of cancer genomic medicine in Japan. Cancer Sci.
112:970–977. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kohno T, Kato M, Kohsaka S, Sudo T, Tamai
I, Shiraishi Y, Okuma Y, Ogasawara D, Suzuki T, Yoshida T and Mano
H: C-CAT: The national datacenter for cancer genomic medicine in
Japan. Cancer Discov. 12:2509–2515. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Riely GJ, Kris MG, Rosenbaum D, Marks J,
Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W, et al: Frequency
and distinctive spectrum of KRAS mutations in never smokers with
lung adenocarcinoma. Clin Cancer Res. 14:5731–5734. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Modest DP, Ricard I, Heinemann V,
Hegewisch-Becker S, Schmiegel W, Porschen R, Stintzing S, Graeven
U, Arnold D, von Weikersthal LF, et al: Outcome according to KRAS-,
NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled
analysis of five randomized trials in metastatic colorectal cancer
by the AIO colorectal cancer study group. Ann Oncol. 27:1746–1753.
2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Halbrook CJ, Lyssiotis CA, Pasca di
Magliano M and Maitra A: Pancreatic cancer: Advances and
challenges. Cell. 186:1729–1754. 2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yang Y, Zhang H, Huang S and Chu Q: KRAS
mutations in solid tumors: Characteristics, current therapeutic
strategy, and potential treatment exploration. J Clin Med.
12(709)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lutsik P, Baude A, Mancarella D, Öz S,
Kühn A, Toth R, Hey J, Toprak UH, Lim J, Nguyen VH, et al: Globally
altered epigenetic landscape and delayed osteogenic differentiation
in H3.3-G34W-mutant giant cell tumor of bone. Nat Commun.
11(5414)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Phillips JJ, Gong H, Chen K, Joseph NM,
van Ziffle J, Jin LW, Bastian BC, Bollen AW, Perry A, Nicolaides T,
et al: Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic
pleomorphic xanthoastrocytoma without BRAF p.V600E mutation. Acta
Neuropathol. 132:757–760. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Isaacson AL, Guseva NV, Bossler AD and Ma
D: Urothelial carcinoma with an NRF1-BRAF rearrangement and
response to targeted therapy. Cold Spring Harb Mol Case Stud.
5(a003848)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chang KTE, Tay AZE, Kuick CH, Chen H,
Algar E, Taubenheim N, Campbell J, Mechinaud F, Campbell M, Super
L, et al: ALK-positive histiocytosis: an expanded clinicopathologic
spectrum and frequent presence of KIF5B-ALK fusion. Mod Pathol.
32:598–608. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kashima J, Yoshida M, Jimbo K, Izutsu K,
Ushiku T, Yonemori K and Yoshida A: ALK-positive histiocytosis of
the breast: A clinicopathologic study highlighting spindle cell
histology. Am J Surg Pathol. 45:347–355. 2021.PubMed/NCBI View Article : Google Scholar
|