Introduction
The most common neorodegenerative disorders, namely
Alzheimer's disease (AD) and Parkinson's disease (PD), are
prevalent in ~1% of individuals aged 60 years and older (1). Their etiologies remain uncertain, though
there are a number of established pathogenic factors, including
oxidative stress, neural apoptosis, mitochondrial dysfunction,
excitotoxicity, impairment of the ubiquitin-proteasome system and
inflammation (2,3). Neuroprotective therapy has been
suggested to prevent disease progression by inhibiting the action
of pathogenic factors, for instance, by reducing the production of
reactive oxygen species (ROS) (4).
Overproduction of ROS may cause oxidative damage to biomolecules
and subsequently DNA damage, ultimately leading to the development
of neurodegenerative diseases. According to studies on PD,
inhibitors of type B monoamine oxidase (MAO), including selegiline
[also known as (−)-deprenyl] and rasagiline, are among the most
promising neuroprotective agents identified to date (5–7). MAO
exists in two forms, MAO-A and -B. The catalytic activity of these
enzymes generates H2O2 and nitrogen species,
which are toxic products that may cause oxidative damage to
mitochondrial DNA (mtDNA) and thus have potential implications for
apoptosis, aging and neurodegenerative processes (8). The MAO-B inhibitor, selegiline, is
typically recommended as a first-line treatment for PD and has been
demonstrated to possess neuroprotective functions (9); notably, this inhibitor protected
neuronal cells against induced cell death in cellular and animal
models (10,11). The neuroprotective functions of
selegiline have been attributed to stabilization of the
mitochondria, to the prevention of death signaling processes, and
to upregulation of the anti-apoptotic B-cell lymphoma 2 (Bcl-2)
protein family and neurotrophic factors (10,11).
Previous studies have also indicated that MAO inhibitors suppress
H2O2-induced oxidative stress and attenuate
the induced cell injury by promoting expression of the Bcl-2 family
(10,11). Unlike other drugs with neuroprotective
properties, selegiline and its metabolites are able to cross the
blood-brain barrier, following which they exhibit highest
accumulation in the thalamus, basal ganglia, mesencephalon and
cingulate gyrus (12,13).
As a follow-up to previous research by our group
(14,15), the present study investigated the
in vitro therapeutic effects of selegiline on the apoptosis
and survival of hippocampus-derived rat neural stem cells (NSCs)
treated with hydrogen peroxide, namely through MTT and terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) assays and acridine orange/ethidium bromide staining, along
with reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) to determine the expression of heat shock protein 4
(Hspa4) and Bcl-2.
Materials and methods
Isolation and expansion of NSCs
NSCs were isolated from the hippocampus of 5
neonatal Wistar rats (10 days old) purchased from the the Razi
Vaccine and Serum Research Institute (Karaj, Iran) using a
neurosphere assay as described previously (16). Prior to cell isolation, the rats were
housed under a 12-h light/dark cycle at 24°C and 30–35% humidity
with food and water available ad libitum. NSCs were
collected from the hippocampus of the rats under anesthesia with
ketamine (100 mg/kg) and xylazine (10 mg/kg; intraperitoneal
injection). The dissected hippocampi were washed in
phosphate-buffered saline (PBS) supplemented with 4.5 g/l glucose
solution and then centrifuged for 5 min at 1,600 × g and 4°C. The
collected tissues in the pellet were dissociated for 30 min at room
temperature (RT) using a digestion mixture of 2.5 U/ml papain
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), 40 U/ml dispase II
(Sigma-Aldrich; Merck KGaA) and 400 U/ml accutase (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), with mixing of
the solution every 10 min. Subsequently, the cell mixture was
passed through a 70-µm cell strainer and then centrifuged for 5 min
at 800 × g and 4°C. Following centrifugation, the pellets were
resuspended in 1 ml Dulbecco's modified Eagle's medium F-12
(DMEM/F12; Invitrogen; Thermo Fisher Scientific, Inc.). The
isolated cells were seeded in a 25-cm2 non-adherent
plastic flask (105 cells/ml) in DMEM/F12 supplemented
with 2% B27 supplement (Gibco; Thermo Fisher Scientific, Inc.), 20
ng/ml basic fibroblast growth factor (bFGF; Invitrogen; Thermo
Fisher Scientific, Inc.), 20 ng/ml epidermal growth factor (EGF;
Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
and 100 mg/ml streptomycin and incubated at 37°C in 5% CO2 for 1–2
weeks to enable neurosphere formation. The medium and growth
factors were replenished every 2 days. Following this, floating
neurospheres were collected by centrifugation at 300 × g for 5 min
at RT. They were then dissociated enzymatically using trypsin-EDTA
(0.25%) and mechanically (by pipetting) to single cells. The cells
were then suspended in DMEM/F12 supplemented with 2% B27, 20 ng/ml
bFGF, 20 ng/ml EGF and 5% fetal bovine serum (Sigma-Aldrich; Merck
KGaA) for 1 week at 37°C and 5% CO2 in 6-well adherent plates
(5×106 cells/well) coated with poly-l-lysine
(Sigma-Aldrich; Merck KGaA) and passaged up to three times. Nestin
as a neural stem/precursor cell marker (17) was evaluated immunocytochemically. The
present study adhered to institutional guidelines for the care and
use of laboratory animals, and all experimental procedures were
reviewed and approved by the Ethics Committee for the Use of
Experimental Animals at Tarbiat Modares University (Tehran,
Iran).
Selegiline cell treatments
H2O2 was used to induce oxidative stress, and was
prepared from 30% stock solution prior to each experiment.
Third-passage NSCs were cultured in 96-well plates (105
cells/well) in DMEM/F12 medium supplemented with 2% B27, 20 ng/ml
bFGF and 20 ng/ml EGF. The NSCs were then incubated at 37°C in 5%
CO2 with 125 µM H2O2 for 30 min. To evaluate the neuroprotective
effects of selegiline, cells were pretreated with different
concentrations of selegiline (Sigma-Aldrich; Merck KGaA; 0, 10, 20,
30 and 40 µM) 48 h prior to the H2O2 treatment. NSCs without
selegilin treatment (0 µM) were used as a control group. The
control cells were cultured in the above DMEM/F12 medium for 48 h
at 37°C, then treated with 125 µM H2O2 for 30 min.
Immunostaining
The hippocampus-derived NSCs were cultured on cover
slides (5×103 cells/slide) in the supplemented DMEM/F12
(2% B27, 20 ng/ml bFGF and 20 ng/ml EGF) for 48 h at 37°C in 5%
CO2, then fixed in 3% paraformaldehyde for 20 min at RT. The cells
were then permeabilized in 100% methanol for 30 min at RT to enable
antibody-antigen interaction. For immunofluorescence, the cells
were incubated with anti-nestin monoclonal antibodies, (1:200;
ab6142; Abcam, Cambridge, UK) for 2 h at 4°C, then with fluorescein
isothiocyanate-conjugated rabbit anti-mouse antibody (1:300;
ab6724; Abcam) for 2 h at RT. Nuclei were counterstained with
ethidium bromide for 1 min at RT to visualize the nuclei. Following
staining, cells were examined using an inverted fluorescence
microscope and the number of immunopositive cells were counted in a
minimum of 100 cells per experiment. Corresponding negative
controls were established using secondary antibodies without
primary antibody incubation, to exclude nonspecific binding of
secondary antibody to the sample.
Viability assay
Cell viability was evaluated using an MTT bromide
assay, as described previously (18).
Cells were treated with 125 µM H2O2 and different concentrations of
selegiline 48 h before the H2O2 treatment. The treated cells were
incubated with 1 mg/ml MTT (Sigma Aldrich; Merck KGaA) for 4 h,
after which the culture medium (DMEM/F12 with 2% B27, 20 ng/ml bFGF
and 20 ng/ml EGF) was removed and 100 µl dimethyl sulfoxide was
added to each well to dissolve the formazan crystals. The quantity
of formazan dissolved was quantified from absorbance at 570 nm
(A570) using a microplate ELISA reader, and relative cell viability
(%) was calculated as follows: (A570 of treated samples/A570 of
controls) ×100 (19,20).
TUNEL detection of apoptotic
cells
Following treatment, the NSCs were fixed with 4%
paraformaldehyde in PBS for 30 min at RT. The cells were subjected
to a TUNEL assay using an In-Situ Cell Death Detection kit
(Roche Applied Science, Penzberg, Germany) according to the
manufacturer's instructions. TUNEL-positive cells were labeled
using diaminobenzidine as the chromogen for 3–7 min at RT, and
counterstained with hematoxylin for 5 min at RT. The percentage of
TUNEL-positive cells was assessed using an Olympus phase contrast
fluorescence microscope (Olympus Corporation, Tokyo, Japan) in five
randomly selected fields for each well.
Acridine orange/ethidium bromide
staining
Necrotic morphological changes in the treated cells
were assessed by acridine orange/ethidium bromide staining.
Following the selegiline and H2O2 treatments, the NSCs were washed
with PBS buffer and fixed with 4% paraformaldehyde for 15 min at
RT, then stained with acridine orange/ethidium bromide
(Sigma-Aldrich; Merck KGaA; 100 µg/ml of each) for 5 min at RT. The
number of necrotic cells, identified by orange/yellow cytoplasmic
staining and by non-condensed chromatin and/or non-fragmented
nuclei (21), were counted in a total
of 200 cells. The cells were observed using a fluorescence
microscope.
RT-qPCR
RT-qPCR was performed with cDNA obtained from the 0
(control) and 20 µM selegiline groups following induced oxidative
stress. A total of 1,000 ng purified RNA obtained from cultured
cells with TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) was
used to synthesize 20 µl cDNA using a RevertAid™ First Strand cDNA
Synthesis kit (Fermentas, Germany) according to the manufacturer's
instructions. The cDNA was used to quantify Bcl-2 and Hspa4 mRNA
levels, with β2-microglobulin (B2M) used as an internal control for
normalization. The primer sequences of all primers used are listed
in Table I. The PCR reaction was
performed in a 25-µl final reaction volume [containing forward and
reverse primers (200 nM each), cDNA (0.5 µl),
SYBR®-Green I (12.5 µl; Fermentas; Thermo Fisher
Scientific, Inc.) and nuclease-free water up to final volume] for
40 cycles at 95°C for 15 sec followed by 60°C for 1 min. Relative
changes in target mRNA levels were determined using the ΔΔCq method
(22).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | GenBank accession
no. | Forward, 5′-3′ | Reverse, 5′-3′ |
|---|
| B2M | NM_012512.2 |
CTTGCCATTCAGAAAACTCC |
CATCGGTCTCGGTGGGTG |
| Bcl-2 | NM_016993 |
ACGGTGGTGGAGGAACTCTTCAGG |
AATCAAACAGAGGTCGCATGCTGG |
| Hspa4 | NM_153629 |
GAGTGCGAATGCTTCAGACCTCCC |
CGTGTGGCTCCACCAACTATCTCC |
Statistical analysis
Data analysis was performed using SPSS 15.0 software
(SPSS, Inc., Chicago, IL, USA). All data are presented as the mean
± standard error of the mean from 5 independent experiments. To
compare differences between the means of multiple groups, one-way
analysis of variance followed by Tukey's post hoc test was used.
P<0.05 was considered to indicate statistical significance.
Results
Generation of NSCs
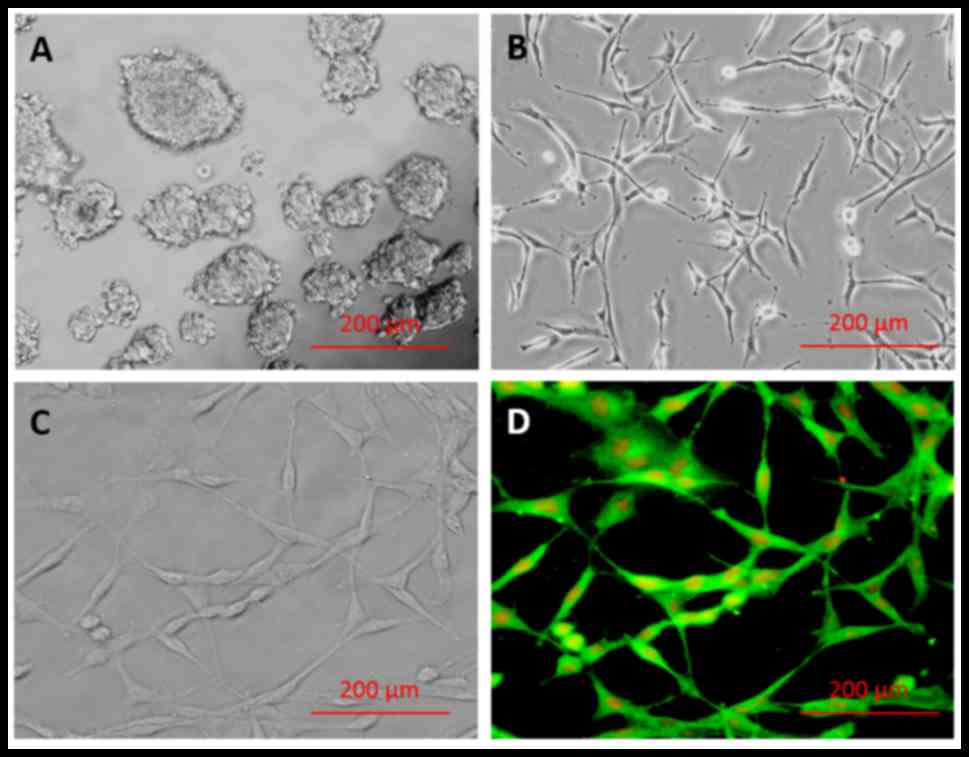
The results of the primary culture of
hippocampus-derived NSCs are presented in Fig. 1A-D. Following NSC isolation and
culture, single cells were observed to form primary neurospheres by
day 7 (Fig. 1A). Secondary
neurospheres originated from the primary neurospheres, and a
homogeneous adherent NSC population was obtained after 3 passages
(Fig. 1B). The majority of isolated
cells (98.71±0.29%) exhibited positive staining for nestin marker
(Fig. 1D), indicating successful
isolation of NSCs, and negative controls incubated in the absence
of primary antibody confirmed the specificity of labeling (data not
shown).
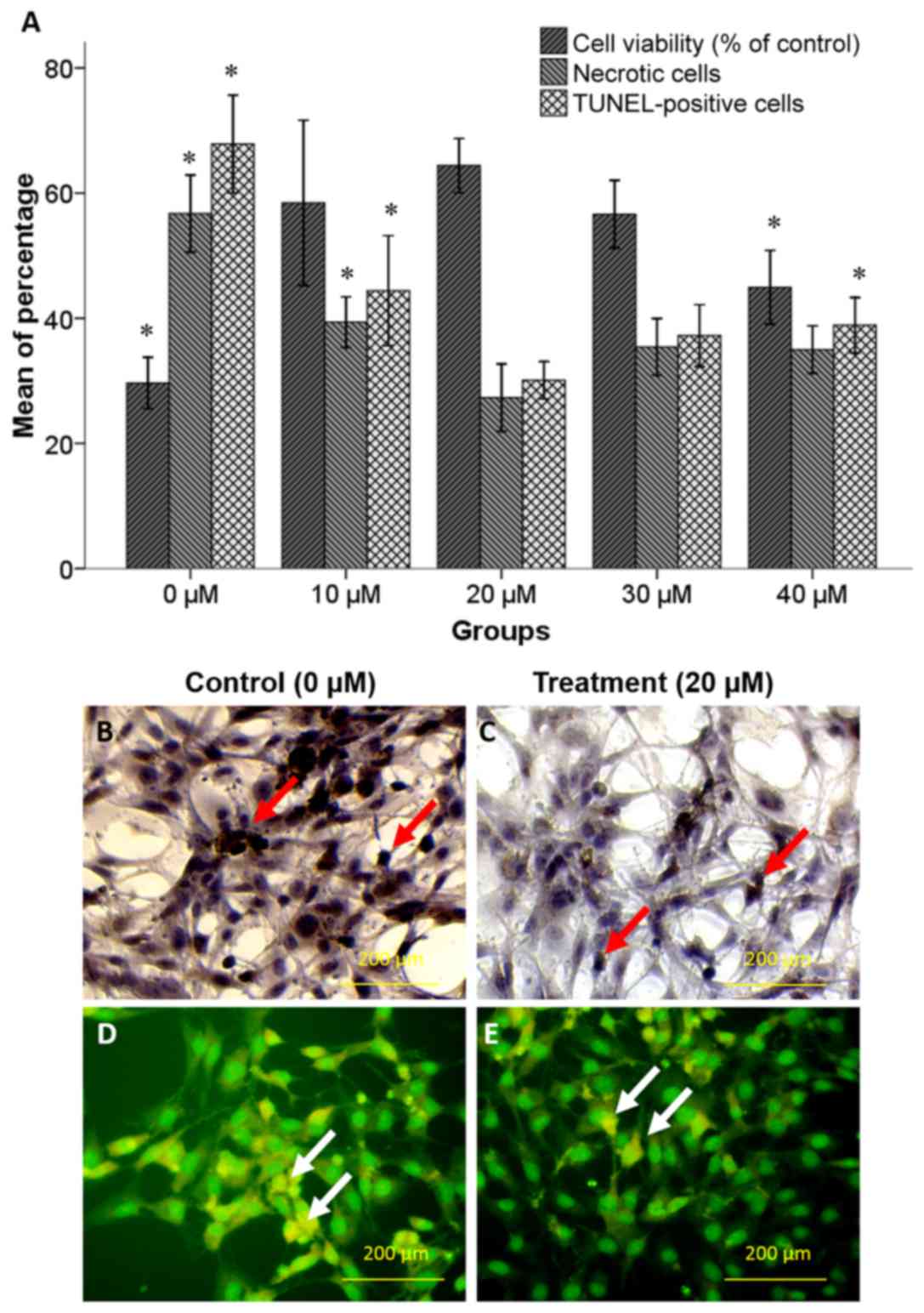
Neuroprotective effect of selegiline
on NSC viability
An MTT assay was performed to determine the
neuroprotective effect of increasing concentrations of selegiline
(0–40 µM) against oxidative stress induced by 125 µM H2O2. The
attained values were normalized based on values of NSCs without any
treatment (negative control group). As depicted in Fig. 2A, pretreatment with 20 µM selegiline
for 48 h caused an increase in the percentage of viable cells
(64.4±2.17%) compared with the 0 (29.66±2.04%; P<0.05) and 10 µM
(58.44±6.60%) selegiline groups. However, further increases in
selegiline concentration caused a decline in cell viability
compared with the 20 µM selegiline group, which was deemed to be
significant for 40 µM (44.94±2.94%; P<0.05).
Neuroprotective effect of selegiline
on NSC survival
A TUNEL assay and acridine orange/ethidium bromide
staining were performed to assess the pro-survival effects of
selegiline in hippocampus-derived NSCs treated with increasing
concentrations of selegiline for 48 h followed by H2O2 exposure.
The TUNEL and necrotic cell staining indicated that H2O2 induced
apoptotic and necrotic cell death (Fig.
2A-E). As depicted in Fig. 2A,
pretreatment with 20 µM selegiline caused significant decreases in
the percentages of apoptotic (30.10±1.48%) and necrotic
(27.32±2.68%) cells compared with the 10 µM selegiline group
(44.4±4.39 and 39.37±2.01%, respectively; P<0.05) and the 0 µM
(control) group (67.84±3.91 and 59.74±3.07%, respectively;
P<0.05). However, further increases in selegiline concentration
caused increased rates of cell apoptosis and necrosis compared with
the 20 µM selegiline group, which was deemed to be significant at
40 µM on cell apoptosis (33.89±2.21%; P<0.05).
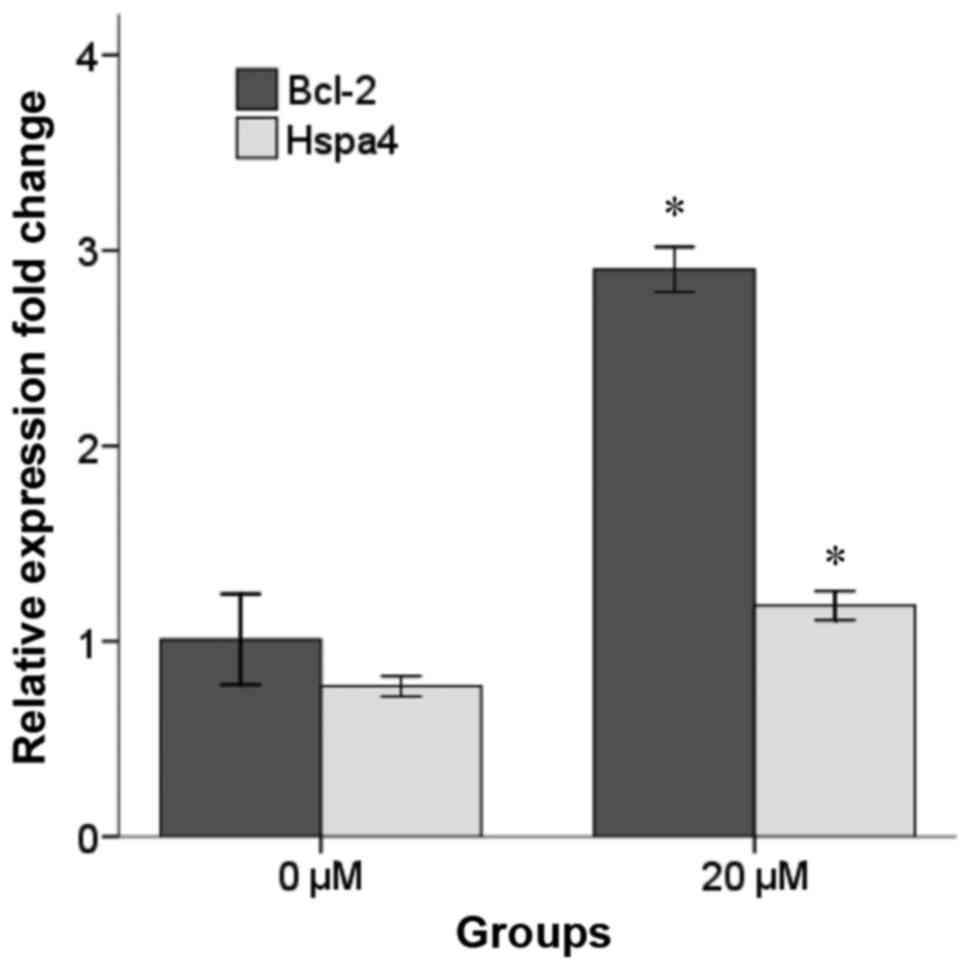
Effect of selegiline on Bcl-2 and
Hspa4 expression
Increased levels of Bcl-2 and Hspa4 mRNA in
H2O2-induced NSCs pretreated with 20 µM selegiline for 48 h were
confirmed by RT-qPCR (Fig. 3).
Notably, following normalization to the expression of B2M, it was
observed that the mRNA levels of Bcl-2 and Hspa4 were significantly
increased in the 20 µM selegiline group (2.90±0.05 and 1.18±0.03,
respectively) compared with the control group (1.01±0.11 and
0.77±0.02, respectively; P<0.05).
Discussion
In the present study, it was demonstrated that
selegiline increased Bcl-2 and Hspa4 mRNA expression in
hippocampus-derived NSCs, and protected the cells against
H2O2-induced apoptosis and necrosis. It has
previously been reported that free radicals serve a causal role in
biomolecular damage and neuronal cell death following oxidative
stress (2,23). In neurodegenerative disorders, a
reduction of oxidative stress may attenuate apoptotic cell death
and disease progression (24). MAO
serves important roles in the generation of
H2O2 and nitrogen species, and in apoptosis,
aging and neurodegenerative processes (8,25).
Furthermore, increased levels of MAO-B mRNA and enzymatic activity
have been observed in neurodegenerative diseases including PD
(26).
In vitro and in vivo experiments have
demonstrated that selegiline is a potent inhibitor of MAO-B and
also enhances the synthesis of neurotrophic factors including glial
cell-derived neurotrophic factor and brain-derived neurotrophic
factor (9,27,28). As a
selective MAO-B inhibitor, selegiline may be used as an anti-PD
drug to exert antioxidant and anti-apoptotic effects (29,30). It
has also been indicated that selegiline decreases oxidative stress
and cell death induced by 1-methyl-4-phenylpyridinium
(MPP+), an inducing agent of PD (31). However, the protective effect of
selegiline against MPP+-induced neuronal cell
degeneration may be opposing dependent on concentration; while
micromolar to submillimolar doses of selegiline promoted increased
cell viability, concentrations of selegiline greater than 1 mM
induced a decrease in cell viability in the MPP+-treated
cells (32). This opposing effect of
selegiline regarding anti-apoptotic activity has also been
demonstrated in A-2058 human melanoma cell culture, in which
selegiline at a concentration range of
10−7−10−3 M caused significant inhibition of
apoptosis, while treatment 10−3 M selegiline caused 50%
apoptosis after treatment for 72 h (29). The current results are in accordance
with these previous studies.
The MAO-B inhibitors, rasagiline and selegiline,
protect neuronal cells through upregulation of the pro-survival
protein Bcl-2 and neurotrophic factors (33). In the present study, increases in the
mRNA levels of Bcl-2 and Hspa4 were identified in hippocampal NSCs
following treatment with 20 µM selegiline for 48 h. The Bcl-2
protein family controls the release of cytochrome c
(Cyt-c) from the mitochondria and consists of pro-apoptotic
(Bcl-2-associated X protein, Bcl-2 homologous antagonist/killer,
BH3 interacting-domain death agonist, Bcl-2-associated death
promoter) and anti-apoptotic [Bcl-2, Bcl-extra large (Bcl-xL)]
members (34). The primary function
of anti-apoptotic Bcl-2 is to suppress mitochondrial Cyt-c
release, by regulating physiological membrane permeability,
Ca2+ release and oxidative stress in the mitochondria
(35,36). Specific regions of Bcl-2, namely the
Bcl-2 homology-3 and −4 domains, exert anti-apoptotic effects in
in vivo and in vitro models (37,38).
Additionally, overexpression of Bcl-2 protein has been documented
to reduce tissue damage in animal models of PD (39,40) and
ischemia (41,42), and protects cells against apoptosis
(43). Furthermore, Bcl-2
overexpression prevented Cyt-c release following
peroxynitrite-induced apoptotic signaling, indicating the potential
of Bcl-2 as a neuroprotective agent (44). In a previous study, selegiline
increased the mRNA and protein levels of Bcl-2 and Bcl-xL in
SH-SY5Y cells (45), but not in
MAO-B-containing Caco-2 and U118MG cells (46,47). These
studies are consistent with present findings regarding the
significant upregulation of Bcl-2 and improved survival of NSCs in
response to 20 µM selegiline.
Interestingly, it has been reported that Hspa4 may
decrease the protein degradation and increase the stability of
Bcl-2 during oxidative stress, and that an association may exist
between Bcl-2 and the anti-apoptotic effect of Hspa4 (48). In consideration of the increases in
Bcl-2 and Hspa4 mRNA and the potential role of Bcl-2 in the
selegiline-treated group, the current study indicated that these
factors may have been associated with the decreased percentages of
necrotic and TUNEL-positive cells decreased in the 20 µM selegiline
group compared with the 0 (control) and 10 µM groups.
In conclusion, the current data suggested that
selegiline is effective in protecting NSCs against oxidative
stress. Therefore, selegiline may be considered as a drug candidate
for neurological disorders in which oxidative stress serves an
important role in pathogenesis.
Acknowledgements
The authors would like to thank Dr Saeid Hashamein
from the Stem Cell Laboratory of Islamic Azad University for
assisting with the research. The study was partly funded by Islamic
Azad University, Ardabil Branch (grant no. 91.502).
References
|
1
|
DeMaagd G and Philip A: Parkinson's
disease and its management: Part 1: Disease entity, risk factors,
pathophysiology, clinical presentation, and diagnosis. PT.
40:504–532. 2015.
|
|
2
|
Uttara B, Singh AV, Zamboni P and Mahajan
RT: Oxidative stress and neurodegenerative diseases: A review of
upstream and downstream antioxidant therapeutic options. Curr
Neuropharmacol. 7:65–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo C, Sun L, Chen X and Zhang D:
Oxidative stress, mitochondrial damage and neurodegenerative
diseases. Neural Regen Res. 8:2003–2014. 2013.PubMed/NCBI
|
|
4
|
Sarkar S, Raymick J and Imam S:
Neuroprotective and therapeutic strategies against Parkinson's
disease: Recent Perspectives. Int J Mol Sci. 17:172016. View Article : Google Scholar
|
|
5
|
Nayak L and Henchcliffe C: Rasagiline in
treatment of Parkinson's disease. Neuropsychiatr Dis Treat.
4:23–32. 2008.PubMed/NCBI
|
|
6
|
Riederer P and Laux G: MAO-inhibitors in
Parkinson's disease. Exp Neurobiol. 20:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonneh-Barkay D, Ziv N and Finberg JP:
Characterization of the neuroprotective activity of rasagiline in
cerebellar granule cells. Neuropharmacology. 48:406–416. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu D, Johnson C, Johnson S, Tazik S and Ou
XM: The neuroprotective effect of antidepressant drug via
inhibition of TIEG2-MAO B mediated cell death. Drug Discov Ther.
2:289–295. 2008.PubMed/NCBI
|
|
9
|
Zhao Q, Cai D and Bai Y: Selegiline
rescues gait deficits and the loss of dopaminergic neurons in a
subacute MPTP mouse model of Parkinson's disease. Int J Mol Med.
32:883–891. 2013.PubMed/NCBI
|
|
10
|
Hara MR, Thomas B, Cascio MB, Bae BI,
Hester LD, Dawson VL, Dawson TM, Sawa A and Snyder SH:
Neuroprotection by pharmacologic blockade of the GAPDH death
cascade. Proc Natl Acad Sci USA. 103:pp. 3887–3889. 2006;
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maruyama W, Akao Y, Carrillo MC, Kitani K,
Youdium MB and Naoi M: Neuroprotection by propargylamines in
Parkinson's disease: Suppression of apoptosis and induction of
prosurvival genes. Neurotoxicol Teratol. 24:675–682. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lecht S, Haroutiunian S, Hoffman A and
Lazarovici P: Rasagiline - a novel MAO B inhibitor in Parkinson's
disease therapy. Ther Clin Risk Manag. 3:467–474. 2007.PubMed/NCBI
|
|
13
|
Fowler JS, Volkow ND, Logan J, Schlyer DJ,
MacGregor RR, Wang GJ, Wolf AP, Pappas N, Alexoff D and Shea C:
Monoamine oxidase B (MAO B) inhibitor therapy in Parkinson's
disease: The degree and reversibility of human brain MAO B
inhibition by Ro 19 6327. Neurology. 43:1984–1992. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abdanipour A, Tiraihi T and Delshad A:
Trans-differentiation of the adipose tissue-derived stem cells into
neuron-like cells expressing neurotrophins by selegiline. Iran
Biomed J. 15:113–121. 2011.PubMed/NCBI
|
|
15
|
Abdanipour A and Tiraihi T: Induction of
adipose-derived stem cell into motoneuron-like cells using
selegiline as preinducer. Brain Res. 1440:23–33. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abdanipour A, Sagha M, Noori-Zadeh A,
Pakzad I and Tiraihi T: In vitro study of the long-term cortisol
treatment effects on the growth rate and proliferation of the
neural stem/precursor cells. Neurol Res. 37:117–124. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki S, Namiki J, Shibata S, Mastuzaki Y
and Okano H: The neural stem/progenitor cell marker nestin is
expressed in proliferative endothelial cells, but not in mature
vasculature. J Histochem Cytochem. 58:721–730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gu J, Chi M, Sun X, Wang G, Li M, Liu L
and Li X: Propofol-induced protection of SH-SY5Y cells against
hydrogen peroxide is associated with the HO-1 via the ERK pathway.
Int J Med Sci. 10:599–606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han J, Talorete TP, Yamada P and Isoda H:
Anti-proliferative and apoptotic effects of oleuropein and
hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology.
59:45–53. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abdanipour A, Noori-Zadeh A, Mesbah-Namin
SA, Bakhtiyari S, Nejatbakhsh R and Anarkooli IJ: Di-(2-ethylhexyl)
phthalate-induced hippocampus-derived neural stem cells
proliferation. Cell J. 19:166–172. 2017.PubMed/NCBI
|
|
21
|
Didenko VV, Ngo H and Baskin DS: Early
necrotic DNA degradation: Presence of blunt-ended DNA breaks, 3′
and 5′ overhangs in apoptosis, but only 5′ overhangs in early
necrosis. Am J Pathol. 162:1571–1578. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan JS, Reed A, Chen F and Stewart CN Jr:
Statistical analysis of real-time PCR data. BMC Bioinformatics.
7:852006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Emerit J, Edeas M and Bricaire F:
Neurodegenerative diseases and oxidative stress. Biomed
Pharmacother. 58:39–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ischiropoulos H and Beckman JS: Oxidative
stress and nitration in neurodegeneration: Cause, effect, or
association? J Clin Invest. 111:163–169. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shih JC, Chen K and Ridd MJ: Monoamine
oxidase: From genes to behavior. Annu Rev Neurosci. 22:197–217.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jakubauskiene E, Janaviciute V, Peciuliene
I, Söderkvist P and Kanopka A: G/A polymorphism in intronic
sequence affects the processing of MAO-B gene in patients with
Parkinson disease. FEBS Lett. 586:3698–3704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Youdim MB, Gross A and Finberg JP:
Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent
inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol.
132:500–506. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagatsu T and Sawada M: Molecular
mechanism of the relation of monoamine oxidase B and its inhibitors
to Parkinson's disease: Possible implications of glial cells. J
Neural Transm Suppl. 71:53–65. 2006. View Article : Google Scholar
|
|
29
|
Magyar K and Szende B: (−)-Deprenyl, a
selective MAO-B inhibitor, with apoptotic and anti-apoptotic
properties. Neurotoxicology. 25:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsao CM, Jhang JG, Chen SJ, Ka SM, Wu TC,
Liaw WJ, Huang HC and Wu CC: Adjuvant potential of selegiline in
attenuating organ dysfunction in septic rats with peritonitis. PLoS
One. 9:e1084552014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
32
|
Chetsawang B, Kooncumchoo P, Govitrapong P
and Ebadi M: 1-Methyl-4-phenyl-pyridinium ion-induced oxidative
stress, c-Jun phosphorylation and DNA fragmentation factor-45
cleavage in SK-N-SH cells are averted by selegiline. Neurochem Int.
53:283–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Inaba-Hasegawa K, Akao Y, Maruyama W and
Naoi M: Rasagiline and selegiline, inhibitors of type B monoamine
oxidase, induce type A monoamine oxidase in human SH-SY5Y cells. J
Neural Transm (Vienna). 120:435–444. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gillies LA and Kuwana T: Apoptosis
regulation at the mitochondrial outer membrane. J Cell Biochem.
115:632–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shacka JJ and Roth KA: Regulation of
neuronal cell death and neurodegeneration by members of the Bcl-2
family: Therapeutic implications. Curr Drug Targets CNS Neurol
Disord. 4:25–39. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwartz PS and Hockenbery DM:
Bcl-2-related survival proteins. Cell Death Differ. 13:1250–1255.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shimizu S, Konishi A, Kodama T and
Tsujimoto Y: BH4 domain of antiapoptotic Bcl-2 family members
closes voltage-dependent anion channel and inhibits apoptotic
mitochondrial changes and cell death. Proc Natl Acad Sci USA.
97:pp. 3100–3105. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Malik JM, Shevtsova Z, Bähr M and Kügler
S: Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL
gene transfer. Mol Ther. 11:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Offen D, Beart PM, Cheung NS, Pascoe CJ,
Hochman A, Gorodin S, Melamed E, Bernard R and Bernard O:
Transgenic mice expressing human Bcl-2 in their neurons are
resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine neurotoxicity. Proc Natl Acad Sci USA. 95:pp.
5789–5794. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Natsume A, Mata M, Goss J, Huang S, Wolfe
D, Oligino T, Glorioso J and Fink DJ: Bcl-2 and GDNF delivered by
HSV-mediated gene transfer act additively to protect dopaminergic
neurons from 6-OHDA-induced degeneration. Exp Neurol. 169:231–238.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cao YJ, Shibata T and Rainov NG:
Liposome-mediated transfer of the bcl-2 gene results in
neuroprotection after in vivo transient focal cerebral ischemia in
an animal model. Gene Ther. 9:415–419. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nuydens R, Dispersyn G, Van Den Kieboom G,
de Jong M, Connors R, Ramaekers F, Borgers M and Geerts H: Bcl-2
protects neuronal cells against taxol-induced apoptosis by inducing
multi-nucleation. Apoptosis. 5:335–343. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nuydens R, Dispersyn G, Van Den Keiboom G,
de Jong M, Connors R, Ramaekers F, Borgers M and Geerts H: Bcl-2
protects against apoptosis-related microtubule alterations in
neuronal cells. Apoptosis. 5:43–51. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Naoi M, Maruyama W, Akao Y, Yi H and
Yamaoka Y: Involvement of type A monoamine oxidase in
neurodegeneration: Regulation of mitochondrial signaling leading to
cell death or neuroprotection. J Neural Transm Suppl. 71:67–77.
2006. View Article : Google Scholar
|
|
46
|
Naoi M, Maruyama W and Inaba-Hasegawa K:
Type A and B monoamine oxidase in age-related neurodegenerative
disorders: Their distinct roles in neuronal death and survival.
Curr Top Med Chem. 12:2177–2188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chiou SH, Ku HH, Tsai TH, Lin HL, Chen LH,
Chien CS, Ho LL, Lee CH and Chang YL: Moclobemide upregulated Bcl-2
expression and induced neural stem cell differentiation into
serotoninergic neuron via extracellular-regulated kinase pathway.
Br J Pharmacol. 148:587–598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jiang B, Liang P, Deng G, Tu Z, Liu M and
Xiao X: Increased stability of Bcl-2 in HSP70-mediated protection
against apoptosis induced by oxidative stress. Cell Stress
Chaperones. 16:143–152. 2011. View Article : Google Scholar : PubMed/NCBI
|