Introduction
Hereditary non-polyposis colorectal carcinoma
(HNPCC) is an autosomal dominant genetic disorder characterized by
a deficiency of DNA mismatch repair proteins (1–3). This
deficiency leads to the formation of various neoplasms including
colorectal carcinoma and malignancies of the endometrium, ovary,
stomach, small bowel, and pancreas. Muir-Torre Syndrome (MTS) is
subtype of HNPCC characterized by the additional presence of
various dermatologic neoplasms including sebaceous adenomas,
sebaceous carcinomas, and keratoacanthomas (1–3). HNPCC
is not traditionally associated with hematologic malignancies
(4).
MDS is defined by the World Health Organization
(WHO) as a group of clonal bone marrow neoplasms characterized by
ineffective hematopoiesis, manifested by morphologic dysplasia in
hematopoietic cells and peripheral cytopenia (5). Therapy-related MDS (t-MDS) is a subtype
of MDS defined by the WHO as a distinct category in the
classification of patients who develop myeloid neoplasms following
cytotoxic therapy (5). Associations
between t-MDS and several germline mutations have been established
but, to our knowledge, no association between t-MDS and MTS has
been reported.
Case report
Patient and methods
A 74-year-old male with MTS presented with a new
onset anemia. Approximately 10 years previously, the patient was
treated at an outside hospital for a variety of malignancies
including gastric adenocarcinoma, colorectal adenocarcinoma,
prostatic adenocarcinoma, and multiple cutaneous neoplasms. The
patient reported undergoing several surgical operations including a
resection of approximately 70% of his stomach for gastric
adenocarcinoma. While specific details of his treatment are
unavailable, the patient reported being treated with fluorouracil
(5-FU) and radiation for his gastric adenocarcinoma.
Initial complete blood count revealed a hemoglobin
of 7.6 g/dl, a reticulocyte count of 2.59, and a platelet count of
95,000 platelets/dl. Given the patient's history and new onset
anemia, an evaluation including a peripheral blood smear and bone
marrow biopsy was performed. FISH analysis and a hematologic panel
of next generation sequencing were performed on the bone marrow
specimen. Written informed consent was obtained from the
patient.
Results
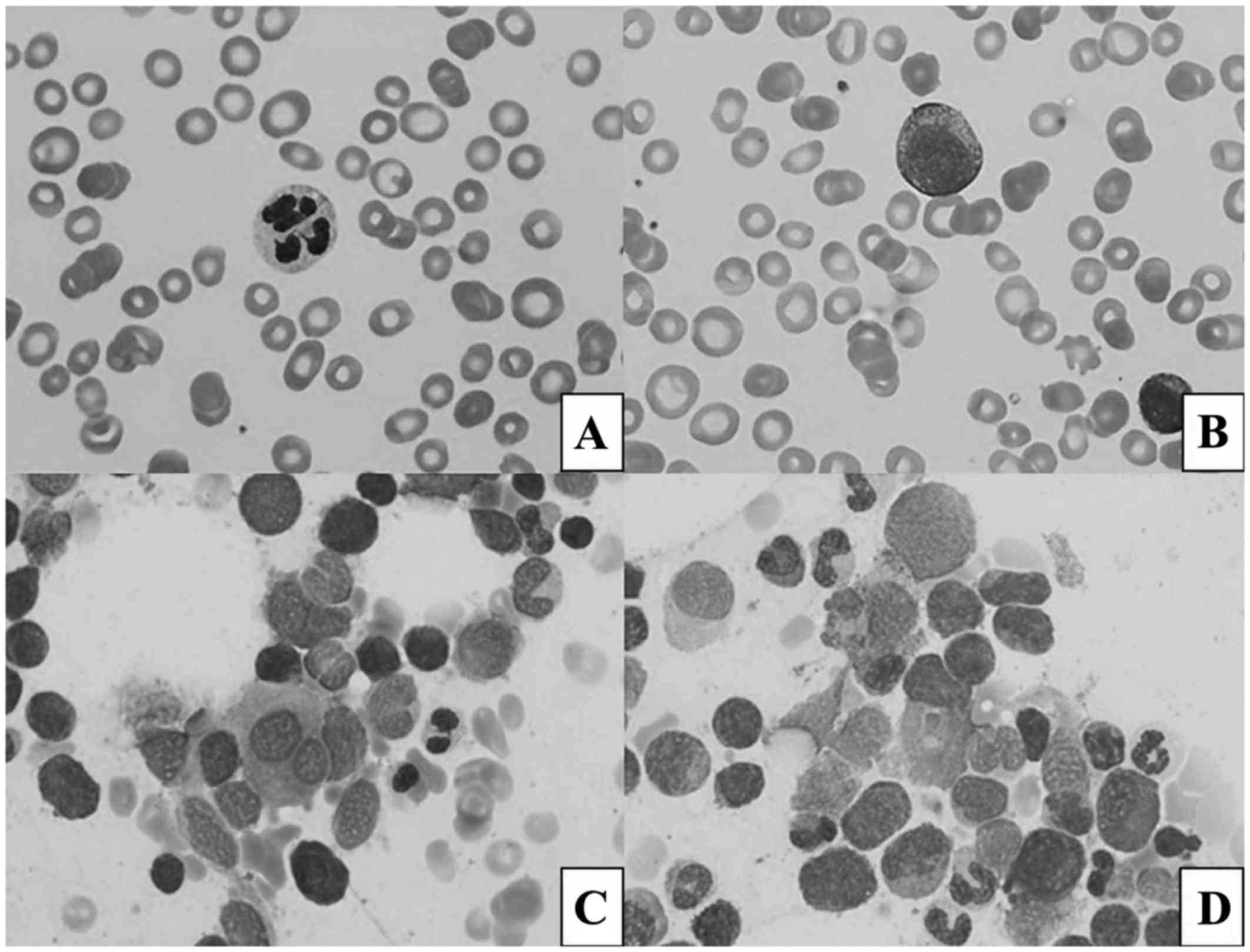
The peripheral blood smear revealed dysplastic,
hypo- and hypersegmented neutrophils and rare blasts (Fig. 1A and B). The bone marrow biopsy
revealed megakaryocytic, erythroid and myeloid dysplasia (Fig. 1C and D) and concurrent flow cytometry
revealed an increased portion of blasts (12%). FISH analysis
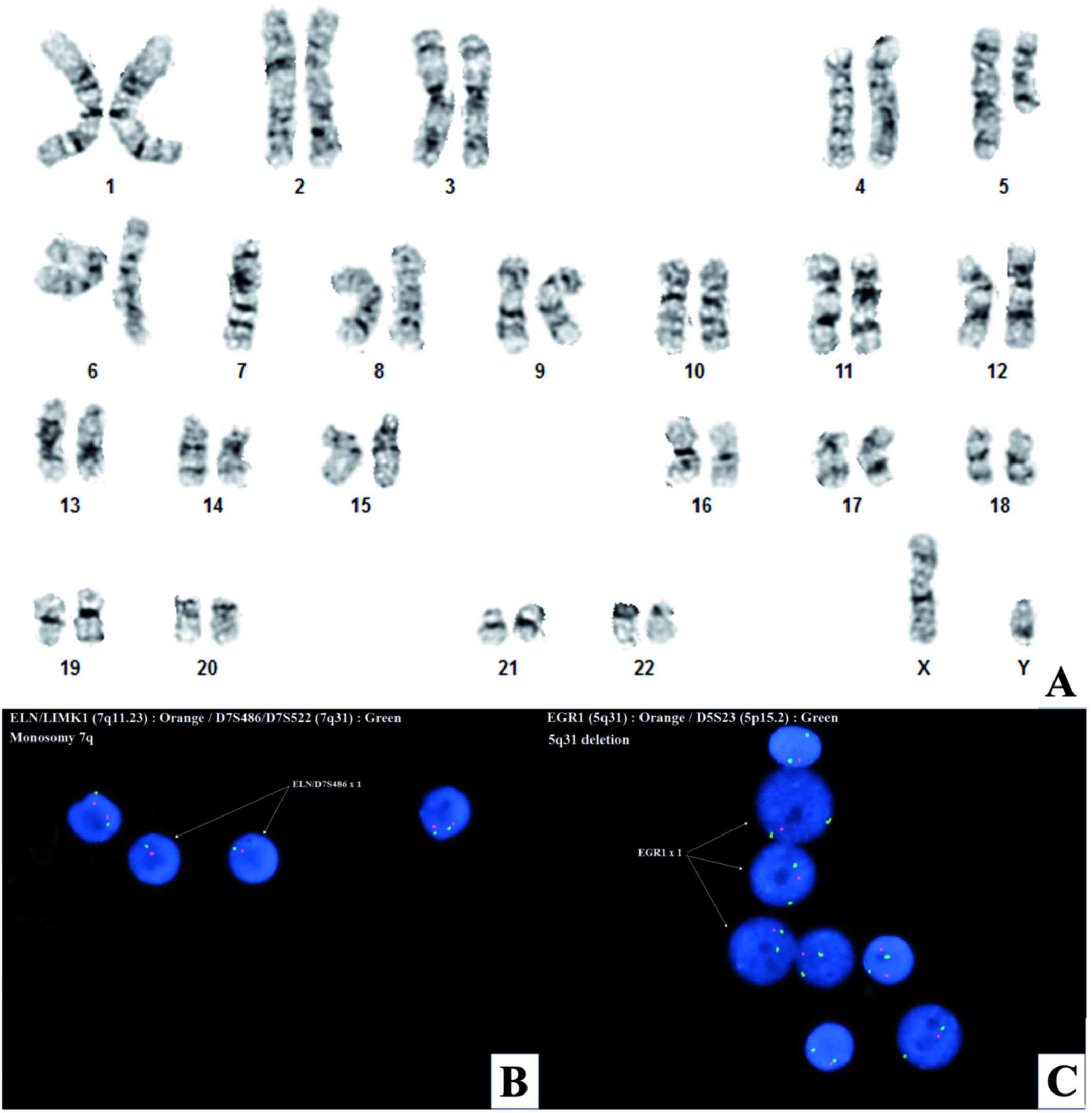
(Fig. 2), revealed 5q31 deletion and
monosomy 7q in ~88% of interphase cells (Fig. 2B and C). G-banded chromosome analysis
demonstrated an interstitial 5q13q33 deletion and monosomy 7 in all
20-metaphase cells examined [45,XY,del(5)(q13q33),-7] (Fig. 2A). A hematologic panel of next
generation sequencing was performed on the bone marrow revealing
mutations in SETBP1 p.G870S and NF1 p.S340Cfs*12. The
patient was diagnosed with refractory anemia with excess blasts
grade II (RAEB-2). A final diagnosis of t-MDS was rendered based on
prior radiation history, multilineage dysplasia and unfavorable
karyotype. The patient's age and existing co-morbidities pose a
significant challenge for the available treatment modalities. The
current patient is an unlikely candidate for stem cell transplant
which is the only potentially curative modality in MDS.
Discussion
Establishing a diagnosis of t-MDS vs. de novo MDS is
a difficult task. In 50 to 70% of t-MDS cases, rearrangements in
the chromosome 5 and/or 7 are present (6). The average time until onset of t-MDS
depends on the type and amount of chemotherapy used. For alkylating
agents and topoisomerase inhibitors the average time until onset of
t-MDS is 5 to 7 years and 2 to 3 years, respectively. Different
chemotherapy agents are associated with different genetic
mutations; alkylating agents being associated with rearrangements
in chromosome 5 and/or 7 (6–10). Our patient presumably developed t-MDS
over 10 years after his treatment with 5-FU and radiation. T-MDS is
traditionally associated with alkylating agents and topoisomerase
inhibitors. However, cases of 5-FU derived t-MDS have also been
reported (11). While HNPCC is not
traditionally associated with hematologic malignancies, mutations
in mismatch repair genes have been detected in hematologic
malignancies and MDS (4,12,13).
Also, there are reports of HNPCC families with neurofibromatosis
like syndromes and increased incidences of hematologic malignancies
(4). A second unrelated primary MDS
in a patient with HNPCC is another possibility. While 10 years is
longer than the average time until onset of MDS and establishing a
definitive etiology may be impossible, the multilineage dysplasia,
complex karyotype and history of chemotherapy and radiation are
indicative of t-MDS.
Next generation sequencing of the marrow indicated
that Muir-Torre syndrome may have played a more important role than
chemotherapy/radiation in the pathogenesis of our patient's t-MDS.
Our patient's bone marrow had a mutation in SETBP1 p.G870S.
As in our patient, SETBP1 p.G870S mutations are associated
with monosomy 7 (14).
Counterintuitively, while monosomy 7 is associated with t-MDS,
SETBP1 p.G870S is not commonly associated with t-MDS
(6–10,15).
Fabiani et al specifically examined SETBP1 mutations
in t-MDS and found only 2.83% (3 of 106) of their therapy related
myeloid neoplasm patient cohort had SETBP1 point mutations
(15). The other mutation detected
in our patient was NF1 p.S340Cfs*12. NF1 mutations
are thought to be causative mutations of neurofibromatosis. Evans
et al found NF1 mutations in 97.08% (166/171) of
familial cases of neurofibromatosis type 1 (16). Interestingly, there are reports of
HNPCC families with increased incidences of hematologic
malignancies and neurofibromatosis like syndromes (4).
Recently a new diagnostic category, ‘myeloid
neoplasms with germline predisposition’, was established by the WHO
(5). The genes currently implicated
in this category are not involved in HNPCC. However, unlike other
hematologic conditions such as chronic myelogenous leukemia, MDS is
a cytogenetically heterogeneous disease (17). As more research is conducted, other
germline mutations such as those involved in HNPCC, maybe
correlated with MDS.
There is an increased frequency of t-MDS in breast
cancer survivors who have germline mutations in breast cancer
susceptibility genes. One recent study found 21% (10/41) of breast
cancer survivors with t-MDS had inherited mutations in at least one
of the following genes: BRCA1, BRCA2, TP53, CHEK2, or
PALB2 (6). T-MDS has also
been reported to occur at an increased frequency in patients with
an inactivating polymorphism in NQ01 gene and the
glutathione S-transferase P1 gene (18,19).
These genes both produce enzymes responsible for metabolizing toxic
byproducts of chemotherapy agents. It has been hypothesized that
certain populations may be susceptible to developing t-MDS due to
an impaired ability to repair DNA damage or an impaired ability to
metabolize chemotherapeutic agents (10).
MTS is caused by an impaired ability to repair DNA
damage, but establishing a correlation with t-MDS may be difficult
due to MTS's low prevalence. Further studies examining the
relationship between t-MDS and MTS are needed. As therapies
continue to improve and life expectancies increase, long term
complications of chemotherapy and radiation such as t-MDS may
become more prevalent. Genetically screening patients undergoing
chemotherapy, and maintaining a central database may allow us to
further elucidate which patients have an increased risk of
developing t-MDS.
References
|
1
|
Lynch HT and de la Chapelle A: Hereditary
colorectal cancer. N Engl J Med. 348:919–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wells K and Wise PE: Hereditary colorectal
cancer syndromes. Surg Clin North Am. 97:605–625. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch HT, Kimberling W, Albano WA, Lynch
JF, Biscone K, Schuelke GS, Sandberg AA, Lipkin M, Deschner EE,
Mikol YB, et al: Hereditary nonpolyposis colorectal cancer (lynch
syndrome I and II). I. Clinical description of resource. Cancer.
56:934–938. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bandipalliam P: Syndrome of early onset
colon cancers, hematologic malignancies & features of
neurofibromatosis in HNPCC families with homozygous mismatch repair
gene mutations. Fam Cancer. 4:323–333. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the world health organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Churpek JE, Marquez R, Neistadt B,
Claussen K, Lee MK, Churpek MM, Huo D, Weiner H, Bannerjee M,
Godley LA, et al: Inherited mutations in cancer susceptibility
genes are common among survivors of breast cancer who develop
therapy-related leukemia. Cancer. 122:304–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Larson RA: Etiology and management of
therapy-related myeloid leukemia. Hematology Am Soc Hematol Educ
Program. 1–459. 2007.PubMed/NCBI
|
|
8
|
Pedersen-Bjergaard J, Andersen MT and
Andersen MK: Genetic pathways in the pathogenesis of
therapy-related myelodysplasia and acute myeloid leukemia.
Hematology Am Soc Hematol Educ Program. 1–397. 2007.PubMed/NCBI
|
|
9
|
Haase D: Cytogenetic features in
myelodysplastic syndromes. Ann Hematol. 87:515–526. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Godley LA and Larson RA: Therapy-related
myeloid leukemia. Semin Oncol. 35:418–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park HJ, Choi JH, Lee KA, Kim HC, Nam YS,
Oh YH and Lee WS: A case of therapy-related acute myeloid leukemia
following 5-fluorouracil chemotherapy. Korean J Intern Med.
27:115–117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hangaishi A, Ogawa S, Mitani K, Hosoya N,
Chiba S, Yazaki Y and Hirai H: Mutations and loss of expression of
a mismatch repair gene, hMLH1, in leukemia and lymphoma cell lines.
Blood. 89:1740–1747. 1997.PubMed/NCBI
|
|
13
|
Maeck L, Haase D, Schoch C, Hiddemann W
and Alves F: Genetic instability in myelodysplastic syndrome:
Detection of microsatellite instability and loss of heterozygosity
in bone marrow samples with karyotype alterations. Br J Haematol.
109:842–846. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hou HA, Kuo YY, Tang JL, Chou WC, Yao M,
Lai YJ, Lin CC, Chen CY, Liu CY, Tseng MH, et al: Clinical
implications of the SETBP1 mutation in patients with primary
myelodysplastic syndrome and its stability during disease
progression. Am J Hematol. 89:181–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fabiani E, Falconi G, Fianchi L, Criscuolo
M, Leone G and Voso MT: SETBP1 mutations in 106 patients with
therapy-related myeloid neoplasms. Haematologica. 99:e152–e153.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Evans DG, Bowers N, Burkitt-Wright E,
Miles E, Garg S, Scott-Kitching V, Penman-Splitt M, Dobbie A,
Howard E, Ealing J, et al: Comprehensive RNA analysis of the NF1
gene in classically affected NF1 affected individuals meeting NIH
criteria has high sensitivity and mutation negative testing is
reassuring in isolated cases with pigmentary features only.
EBioMedicine. 7:212–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haase D, Germing U, Schanz J, Pfeilstöcker
M, Nösslinger T, Hildebrandt B, Kundgen A, Lübbert M, Kunzmann R,
Giagounidis AA, et al: New insights into the prognostic impact of
the karyotype in MDS and correlation with subtypes: Evidence from a
core dataset of 2124 patients. Blood. 110:4385–4395. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Allan JM, Wild CP, Rollinson S, Willett
EV, Moorman AV, Dovey GJ, Roddam PL, Roman E, Cartwright RA and
Morgan GJ: Polymorphism in glutathione S-transferase P1 is
associated with susceptibility to chemotherapy-induced leukemia.
Proc Natl Acad Sci USA. 98:pp. 11592–11597. 2001; View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Larson RA, Wang Y, Banerjee M, Wiemels J,
Hartford C, Le Beau MM and Smith MT: Prevalence of the inactivating
609C->T polymorphism in the NAD(P)H:quinone oxidoreductase
(NQO1) gene in patients with primary and therapy-related myeloid
leukemia. Blood. 94:803–807. 1999.PubMed/NCBI
|