Introduction
Colorectal cancer (CRC) is one of the most prevalent
malignancies and has the third highest mortality rate worldwide.
According to a previous report, >1,880,725 new cases of CRC and
915,880 deaths occurred in 2020 (1). The 5-year relative survival rate for
patients with CRC ranges from 90 to 14% for those diagnosed with
localized to advanced-stage disease (2). Depending on the cancer stage and
tumor location, treatments, including surgery, chemotherapy and
radiotherapy can be used in combination (3). However, the complete removal of total
CRC cells is often not possible. Over 60% of patients with stage II
and III CRC usually undergo adjuvant chemotherapy and/or
radiotherapy; however, these treatments have several side-effects
due to the low cancer-specific selectivity of chemotherapeutic
agents and cytotoxicity. Moreover, a number of patients with CRC
relapse even after neoadjuvant therapy (4,5).
Over the past decade, in order to overcome low specificity and
severe side-effects of classical treatments, molecular targeted
therapies have been developed for personalized medicine (6-8).
Small molecules and mono-clonal antibodies are the major drugs
administered as targeted therapies, including vemurafenib,
cetuximab, trastuzumab and bevacizumab, and are able to suppress
tumor growth, migration and angiogenesis by inhibiting their
specific target molecules (9-12).
Nevertheless, there are still therapeutic limitations, including
long-term toxicity, target gene mutation, drug resistance and
metastasis, as cancer cells may evolve through genetic and
epigenetic clonal diversity (13-16).
Thus, the identification of novel therapeutic targets and the
development of novel approaches to refine or replace existing CRC
chemotherapy are urgently required.
Endothelin (EDN) was first isolated from cultured
porcine aortic endothelial cells, and was identified as the most
potent vasoconstrictor of coronary artery strips (17). In addition, three different EDN
peptides, EDN1, EDN2 and EDN3, have been identified in humans and
other mammalian species (18).
Each EDN peptide is derived from a large precursor of ~200 amino
acids that is first processed by furin endopeptidase, producing an
intermediate product of 38 amino acids. Finally, EDNs are cleaved
by endothelin-converting enzymes to generate the active form of the
EDN peptide (19). The biological
actions of EDN in mammals have been reported to be mediated by EDN
receptors (EDNRs), namely EDNRA and EDNRB. EDNRs belong to the G
protein-coupled receptor (GPCR) family (19). Compared to EDN3, EDN1 and EDN2 bind
to EDNRA with >100-fold higher affinity. By contrast, EDN1, EDN2
and EDN3 bind to EDNRB with equal affinities (20). Specifically, EDNRA mediates
cellular processes by modulating various signaling targets in the
MAPK/ERK and cAMP/protein-kinase A pathways by interacting with
both Gq and Gs/Gi proteins (21-23).
EDN1 and EDNRA are both widely expressed in vascular or
non-vascular tissues and are involved in the regulation of various
physiological processes, including cardiovascular development,
blood pressure regulation, proliferation and migration (24,25).
However, the abnormal activation of EDNRA has been shown to be
associated with several diseases, including pulmonary arterial
hypertension, heart failure and growth retardation. In addition,
EDNRA is overexpressed in several types of cancer, including
prostate, colon, breast and cervical cancer (21,26,27).
In certain studies, increased EDNRA levels stimulated by hypoxia or
microRNAs (miRNAs/miRs) have been shown to lead to the promotion of
cell proliferation, migration, invasion, metastasis and anticancer
drug resistance through G-proteins or β-arrestin downstream
signaling pathways (28,29). However, the detailed regulatory
mechanisms of EDN1 and EDNRA expression have not yet been
completely elucidated and thus warrant further investigation.
In the present study, it was demonstrated that EDN1
and EDNRA promote cell proliferation and migration, and suppress
apoptosis in CRC. The activation of oncogenic signal transducer and
activator of transcription 3 (STAT3) signaling appeared to be
crucial for the concurrent induction of EDN1 and EDNRA expression.
Thus, the findings of the present study elucidate the novel, to the
best of our knowledge, regulatory mechanism of EDN1/EDNRA
expression through the interaction between EDNRA and the STAT3
signaling pathway, leading to the promotion of cell growth and the
suppression of apoptosis by the positive feedback loop of
STAT3-EDN1-EDNRA in CRC. Therefore, the present study indicates
that the EDN1/EDNRA axis may be an emerging therapeutic target for
CRC.
Materials and methods
Cell culture and chemical reagents
The human CRC cell lines, HCT116, SW480, SW620 and
LS174T, were purchased from the Korean Cell Line Bank (KCLB nos.
#10247, #10228, #10227 and #10188, respectively), and cultured in
RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a 5%
CO2 incubator. HT29 cells (KCLB no. #30038), which
originate from AN adenocarcinoma of the rectosigmoid part of the
intestine (30), were cultured in
RPMI-1640 medium supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.). KM12SM cells (KCLB no. #80016) were cultured in
DMEM supplemented with 10% FBS. For STAT3 inhibitor treatment, the
cells were treated with stattic (5 µM; Selleck Chemicals
LLC). For EDNRA inhibitor treatment, the CRC cells were treated
with macitentan (2 or 10 µM; Merck KGaA).
Plasmid construction and
transfection
The coding region of the gene of interest, EDN1 and
EDNRA, was subcloned into the pIRES-EGFP vector (Takara Bio, Inc.).
In summary, EDN1 and EDNRA gene CDS were amplified by PCR using
specific primers (Table SI), and
then inserted into the pIRES-EGFP expression vector. To transfect
the EDN1 and EDNRA plasmids into CRC cell lines, 1×106
cells were plated on a 60 mm dish. After 24 h, 5 µg plasmid
DNA were transfected into the cells using Lipofectamine
2000® reagent (Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Following 24 h of incubation in
a humidified atmosphere of 5% CO2 and at 37°C,
subsequent experiments were carried out.
siRNA transfection
For knockdown experiments, the sequences of siRNAs
(Bioneer Corporation) were used as follows: siRNA for negative
control (siNC) sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and
antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′; siRNA for EDN1
(siEDN1) sense, 5′-CUC GUA GAAG UCU GGU CUA-3′ and antisense,
5′-UAG ACC AGA CUU CUA CGA G-3′; siRNA for EDNRA (siEDNRA) sense,
5′-GCA CUG GUU GGA UGU GUA A-3′ and antisense, 5′-UUA CAC AUC CAA
CCA GUG C-3′; siRNA for STAT3 (siSTAT3) sense, 5′-CAC AUG CCA CUU
UGG UGU UUC AUA A-3′ and antisense, 5′-UUA UGA AAC ACC AAA GUG GCA
UGU G-3′; siRNA for β-arrestin2 (siβ-arr2) sense, 5′-AAG GAC CGC
AAA GUG UUU GUG-3′ and antisense, 5′-CAC AAA CAC UUU GCG GUC
CUU-3′. To transfect the siRNAs, 1×106 CRC cells were
plated on a 60-mm dish. After 24 h, the siRNAs were transfected
into the cells using Lipofectamine RNAiMAX reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
transfected cells were incubated in a humidified atmosphere of 5%
CO2 and at 37°C for 48 h, and then subsequent
experiments were carried out.
Assays of cell growth and migration
CRC cells were seeded into 96-well plates, and cell
viability was measured using the Cell Counting Kit-8 (CCK-8;
Dojindo Laboratories, Inc.) reagent after 6, 24, 48 or 72 h. For
the Transwell migration assay, 3×105 cells were seeded
in the upper chamber (8 µm; Corning Life Sciences) without
FBS medium, and the bottom chambers were filled with 500 µl
medium containing FBS. The migrated cells were fixed with 4%
formaldehyde and 100% methanol at room temperature, and after 24 h,
stained with 0.1% crystal violet (cat. no. V5256, MilliporeSigma)
at room temperature, and counted.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
For total RNA extraction, the NucleoZol reagent
(Macherey-Nagel GmbH) was used following the manufacturer's
instructions. cDNA was synthesized using the PrimeScript RT reagent
kit (cat. no. RR037A, Takara Bio, Inc.). EDN1, EDNRA and GAPDH
expression was quantified by SYBR-Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using gene-specific
oligonucleotide primers (Table
SI) on a StepOne Plus Real-Time System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). qPCR was performed at 95°C for an
initial 3 min followed by 40 cycles of 10 sec at 95°C and 1 min at
59°C. The expression levels of GAPDH were used to normalize
the expression levels of EDN1 and EDNRA. All
reactions were analyzed by the comparative 2−∆∆Cq method
(31).
Western blot analysis
The cells were harvested and lysed using RIPA lysis
buffer (Thermo Fisher Scientific, Inc.) with protease inhibitor
cocktail (Thermo Fisher Scientific, Inc.). The cell lysates were
centrifuged at 12,000 × g at 4°C for 15 min, and a Pierce BCA
Protein Assay kit (cat. no. 23227, Thermo Fisher Scientific, Inc.)
was used for the quantitation of protein lysates. Subsequently, the
lysates were boiled for 10 min. For electrophoresis, 20 µg
protein samples were separated on a 10% polyacrylamide SDS-PAGE gel
and transferred to PVDF membranes (MilliporeSigma). For blocking,
the membranes were incubated with 3% BSA (cat. no. A7906,
MilliporeSigma) in PBS containing 0.1% Tween-20 (cat. no. P1379,
MilliporeSigma) at room temperature for 1 h with agitation. The
membranes were incubated with the primary antibodies [anti-EDNRA
(1:1,000; cat. no. ab85163, Abcam), anti-EDN1 (1:1,000; cat. no.
ab2786, Abcam), anti-phosphorylated (p-) STAT3 (1:1,000; cat. no.
9145, Cell Signaling Technology, Inc.), anti-STAT3 (1:1,000; cat.
no. 12640, Cell Signaling Technology, Inc.), anti-β-actin (1:2,000;
cat. no. AbC-2004, AbClon, Inc.), anti-caspase-3 (1:1,000; cat. no.
9662, Cell Signaling Technology, Inc.), anti-phosphorylated
(p-)p70S6K (1:1,000; cat. no. 9204, Cell Signaling Technology,
Inc.) and anti-p70S6K (1:1,000; cat. no. 9202, Cell Signaling
Technology, Inc.)] at 4°C overnight. The membranes were washed with
PBST and then incubated with a specific secondary antibody, which
was an anti-mouse IgG HRP-linked antibody (1:10,000; cat. no. 7076,
Cell Signaling Technology, Inc.) or anti-rabbit IgG HRP-linked
antibody (1:10,000; cat. no. 7074, Cell Signaling Technology, Inc.)
at room temperature for 1 h. After washing with PBST, the Proteins
were detected by chemiluminescence using an ECL Prime Western
Blotting System (cat. no. RPN2232, Cytiva). The band densities were
determined using ImageJ software (version 1.53k; National
Institutes of Health).
Phosphokinase array
For the phosphokinase assay, the Proteome Profiler
Human Phospho-Kinase Array kit (cat. no. ARY003B, R&D Systems,
Inc.) was used. Briefly, the HCT116 cells were transfected with
negative control siRNA (siNC) or siRNA for EDNRA (siEDNRA). The
cells were lysed with lysis buffer 6 (R&D Systems, Inc.) and
incubated for 30 min on ice, after which the protein lysates were
centrifuged at 14,000 × g for 5 min at 4°C. The BCA assay (cat. no.
23227, Thermo Fisher Scientific, Inc.) was used for the
quantitation of protein lysates. The membranes were incubated with
2 ml of diluted cell lysates on a rocking platform shaker at 4°C
overnight and then incubated with detection antibodies (DAC-A or
DAC-B, provided with the kit by R&D Systems, Inc.). Proteins
were detected by chemiluminescence using an ECL Prime Western
Blotting System (Cytiva, Inc.). Spot densities of the phosphokinase
array were determined by ImageJ software.
Cellular apoptosis assay
For flow cytometric analysis, the HCT116 cells were
seeded and transfected with siRNA or chemotherapeutic agents,
including cisplatin and macitentan, for 48 or 72 h. The cells were
harvested and suspended at 1×105 in 100 µl of PBS
with binding buffer (BD Biosciences). Subsequently, the cells were
stained with 5 µl of Annexin V conjugated to fluorescein
isothiocyanate (FITC) and 5 µl of propidium iodide (PI; BD
Biosciences) at room temperature, for 15 min. Apoptosis analysis
was performed using a FACSVerse flow cytometer (BD Bioscience). For
another cellular apoptosis assay, HCT116 cells were seeded at
1×105 in white 96-well plates. After 24 h, HCT116 cells
were treated with 100 nM ET-1 and 2 µM or 10 µM
macitentan for 6 h at 37°C, and the cells were then equilibrated to
room temperature for 30 min, and 100 µl of Caspase-Glo 3/7
reagent (cat no. G8090, Promega Corporation) was added to each well
and gently mixed by hand tapping. The plate was measured using a
Centro XS3 Luminescence Microplate Reader (Berthold Technologies
GmbH & Co.KG).
Chromatin immunoprecipitation (ChIP)
assay
The STAT3-binding motif in the promoter region of
EDNRA was identified by JASPAR (http://jaspar.genereg.net/). The sequence of the
promoter region was confirmed in the Eukaryotic Promoter Database
(EPD) database (https://epd.epfl.ch/). ChIP was
performed using Dynabeads Protein A and G (Thermo Fisher
Scientific, Inc.). A total of 1×107 cells were
cross-linked with 1% formaldehyde for 10 min at 25°C with continued
agitation, and the crosslinking reaction was stopped by adding
glycine to a final concentration of 0.125 M for 5 min at 25°C with
continued agitation. The cells were then resuspended and lysed for
10 min at 4°C with ChIP lysis buffer. The lysate was sonicated
using an ultrasonicator (Sonics & Materials, Inc.). Fragment
sizes were sized approximately at 250-750 bp. The samples were
diluted with ChIP lysis buffer and pre-cleared with 50 µl of
Dynabeads Protein A and G for 1 h at 4°C. Primary antibodies were
added to the precleared supernatants, and the mixtures were
incubated overnight at 4°C. The antibodies used for the ChIP assay
included anti-STAT3 (dilution 1:100; rabbit polyclonal; cat. no.
9139S; Cell Signaling) and IgG (dilution 1:1,000; cat. no. sc-2357;
Santa Cruz Biotechnology, Inc.). Subsequently, 50 µl of
Dynabeads Protein A and G were added to the samples, and the
mixtures were incubated for 2 h at 4°C. The beads were subsequently
washed with wash buffers (low-salt RIPA, high-salt RIPA, LiCl, and
TE), and the precipitated chromatin was eluted in 100 µl
elution buffer with 0.1 M NaHCO3 (MilliporeSigma) and 1%
SDS (MilliporeSigma) for ≥15 min at 65°C. The chromatin was then
treated with RNase A for 1 h at 37°C, and proteinase K was added to
each sample for 1 h at 65°C. Reverse cross-linking was performed
overnight at 65°C, and DNA was purified using a QIAquick PCR
Purification kit (cat. no. 28104, Qiagen, Inc.). ChIP-PCR assays
were performed using PCR Master Mix (cat. no. 4309155, Thermo
Fisher Scientific, Inc.) and 1% agarose gel (cat. no. 50004, Lonza
Group, Ltd.) electrophoresis.
Plasmid construction for promoter
assay
The EDN1 and EDNRA promoter regions (from −1,000 to
+100 bp) were amplified using the primers listed in Table SI. Subsequently, the PCR products
were purified with a QIAquick PCR Purification kit (Qiagen, Inc.).
To construct the vector, the promoterless pGL4 luciferase vector
(Promega Corporation) was modified. Briefly, the SV40 promoter and
PCR product were ligated with the pGL4.10 basic vector, and the
sequences were then confirmed by Sanger sequencing using RVprimer3
(Promega Corporation).
Promoter assay
HCT116 cells were co-transfected with the
pGL4.10-EDN1 promoter region or PGL4.10-EDNRA promoter region, and
pRL-TK vectors with 50 nM siNC or siRNA for STAT3 (siSTAT3) using
Lipofectamine 2000® (Thermo Fisher Scientific, Inc.).
After 24 h, a luciferase assay was performed using the Dual-Glo
Luciferase Assay System (Promega Corporation) according to the
manufacturer's instructions. Firefly and Renilla luciferase
activities were measured using the Centro XS3 Luminescence
Microplate Reader (Berthold Technologies GmbH & Co.KG).
Tissue microarray
For the tissue microarray, paraffin-embedded slide
glasses, including 29 paired colon tumor/adjacent normal samples,
were purchased from Biochain. For hematoxylin and eosin (H&E)
staining, the tissue sections were deparaffinized, and stained with
hematoxylin (cat. no. S3309, Dako; Agilent Technologies, Inc.) for
1 min at room temperature and eosin (cat. no. CS701, Dako; Agilent
Technologies, Inc.) for 3 min at room temperature. The slices were
dehydrated, and the tissues were then sealed with mounting
solution. Briefly, for the immunohistochemical staining of EDN1 and
EDNRA, tissue sections were deparaffinized, incubated with 3%
H2O2 at room temperature for 10 min, and
washed with PBS three times. The tissues were incubated with 5%
normal goat serum at room temperature for 1 h with anti-EDNRA
(1:200; cat. no. ab117521, Abcam) or anti-EDN1 (1:200; cat. no.
ab2786, Abcam) at 4°C for 12 h. ImmPRESS® HRP
anti-rabbit IgG polymer kit (cat. no. MP-7451, Vector Laboratories,
Inc.) or ImmPRESS® goat anti-mouse IgG polymer kit (cat.
no. MP-7452, Vector Laboratories, Inc.) was added as a secondary
antibody for 1 h, followed by rinsing. Then, 3,3′-diaminobenzidine,
a DAB substrate (cat. no. SK-4105, Vector Laboratories, Inc.), was
added for 2 min at room temperature to develop color.
Analysis of human colon cancer
samples
The Gene Expression database of Normal and Tumor
tissues 2 (GENT2) (http://gent2.appex.kr/gent2/) was analyzed for
EDN1, EDN2, EDN3, EDNRA and
EDNRB expression in various cancer types including breast,
colorectal, and pancreatic cancer. The overall survival data of
EDNRA and EDNRB also was analyzed from GENT2 database with
Kaplan-Meier plots by median cut-off. Publicly available data from
The Cancer Genome Atlas (TCGA) database were analyzed in the
present study. Clinical information and mRNA expression data of
TCGA samples were downloaded from UCSC Xena (http://xena.ucsc.edu). Patient clinical data of colon
cancer were obtained from the TCGA database. Among the colon
adenocarcinoma (COAD) TCGA cohort, 471 tumor and 41 normal colon
samples were analyzed in the present study. The reads per kilobase
of exon per million reads mapped value was used to represent gene
expression levels.
Statistical analysis
All statistical data were conducted using GraphPad
Prism, version 9.0, software package (GraphPad Software). All data
are presented as the mean ± standard deviation, unless otherwise
indicated. Depending on the sample size, the Kolmogorov-Smirnov
test (when n>50) or Shapiro-Wilk test (when n<50) was
performed for the normality test. The unpaired Student's t-test or
Mann-Whitney test was used to perform the analysis of differences
between two groups. For comparison of multiple groups, one-way
ANOVA was performed along with post hoc multiple comparison tests,
including SNK, Dunnett's and Tukey's tests. For correlation
analysis of each gene, the Spearman's correlation analysis was
used. The overall survival from the GENT2 database was analyzed
using the log-rank test. P<0.05 was considered to indicate a
statistically significant difference.
Results
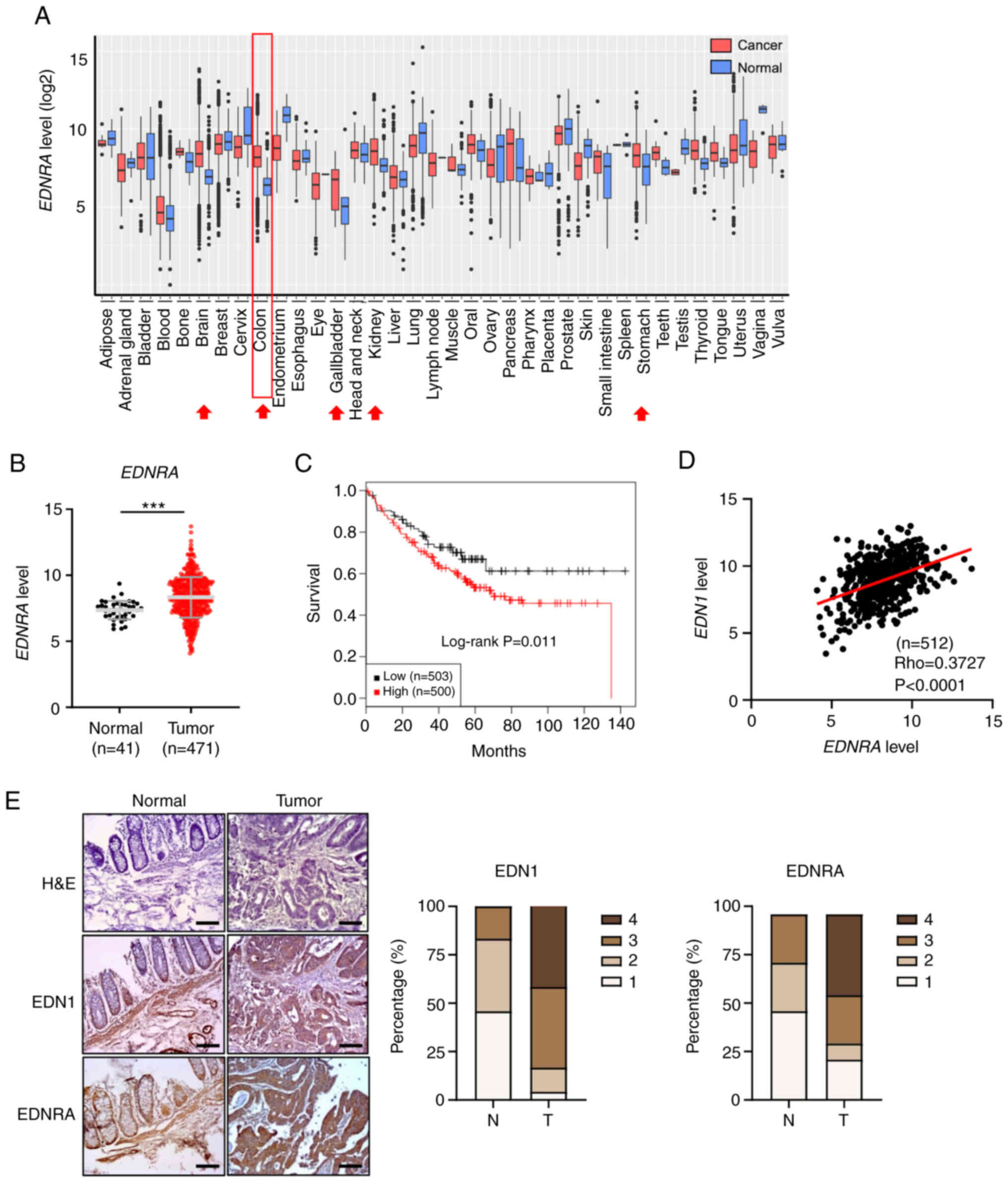
EDNRA expression is upregulated in CRC
and positively correlates with EDN1 levels
Previous research has demonstrated that EDNRA
expression is upregulated in several cancer types, including
bladder and gastric cancer (18).
Since the EDNRA expression level has yet not been clearly
determined in CRC, in the present study, its expression level was
first analyzed using the GENT2 dataset (http://gent2.appex.kr/gent2/) based on microarray data
collected from 36 different cancer types, including breast, skin
and colon cancers. It was observed that EDNRA expression was
markedly upregulated in several cancer types, including brain,
stomach, thyroid, kidney, bone and pancreatic cancers, as well as
CRC (Fig. 1A and Table SII). Similarly, the analysis of an
RNA sequencing dataset from the TCGA-COAD cohort also displayed a
high expression level of EDNRA in CRC samples (Fig. 1B). By contrast, the expression of
the second EDN receptor, EDNRB, was downregulated in CRC tumors
compared with normal tissues (Fig.
S1A and B). Furthermore, it was determined that a high
expression of EDNRA in patients with CRC was associated with a poor
patient survival (Fig. 1C),
whereas no significant difference in overall survival was observed
between patients with increased and decreased EDNRB expression
(Fig. S1C).
Subsequently, the EDNRA ligands, EDN1,
EDN2 and EDN3, from the GENT2 and TCGA datasets were
analyzed. Although the expression of all three ligands was
downregulated in CRC tissues compared to normal tissues (Fig. S2A and B), positive correlations
were determined between the EDN1 and EDNRA or
EDNRB levels (Figs. 1D and
S1D). To further examine the
protein expression of EDN1 and EDNRA, a tissue microarray analysis
was performed, and the increased expression of EDNRA and EDN1 was
detected in the tumor tissues compared to the normal tissues
(Fig. 1E). Therefore, these
results indicated that the interaction between EDNRA and its
ligand, EDN1, may be suggestive of the etiology of CRC.
EDN1/EDNRA regulates the proliferative
and migratory ability of CRC cells
Based on the results of public data analysis,
several functional assays were conducted, in order to elucidate the
effects of EDNRA and EDN1 expression in CRC cells. Firstly, the
endogenous EDNRA mRNA expression levels were confirmed in six CRC
cell lines. Among these, the SW480 and LS174T cells demonstrated
the lowest EDNRA expression levels, whereas the highest EDNRA
expression levels were observed in the HT29 and HCT116 cells
(Fig. S3A). The KM12SM and SW620
cells demonstrated moderate EDNRA expression levels. The
transfection of the EDNRA overexpression vector into SW480 KM12SM
cells with a low EDNRA expression increased the mRNA and protein
levels of EDNRA (Figs. 2A and
S3B). Compared with the control
vector, the EDNRA overexpression vector significantly promoted the
growth and migratory ability of the SW480 and KM12SM cells
(Figs. 2B and C, and S3C and D). The knockdown of EDNRA using
siRNA targeting EDNRA (siEDNRA) decreased the EDNRA mRNA and
protein levels in HCT116 cells (Fig.
2D). Transfection with siEDNRA significantly reduced the growth
of HCT116 cells (Fig. 2E). In
addition, the silencing of EDNRA in HCT116 cells resulted in a
significantly lower number of migrated cells than that in the
negative control cells (Fig.
2F).
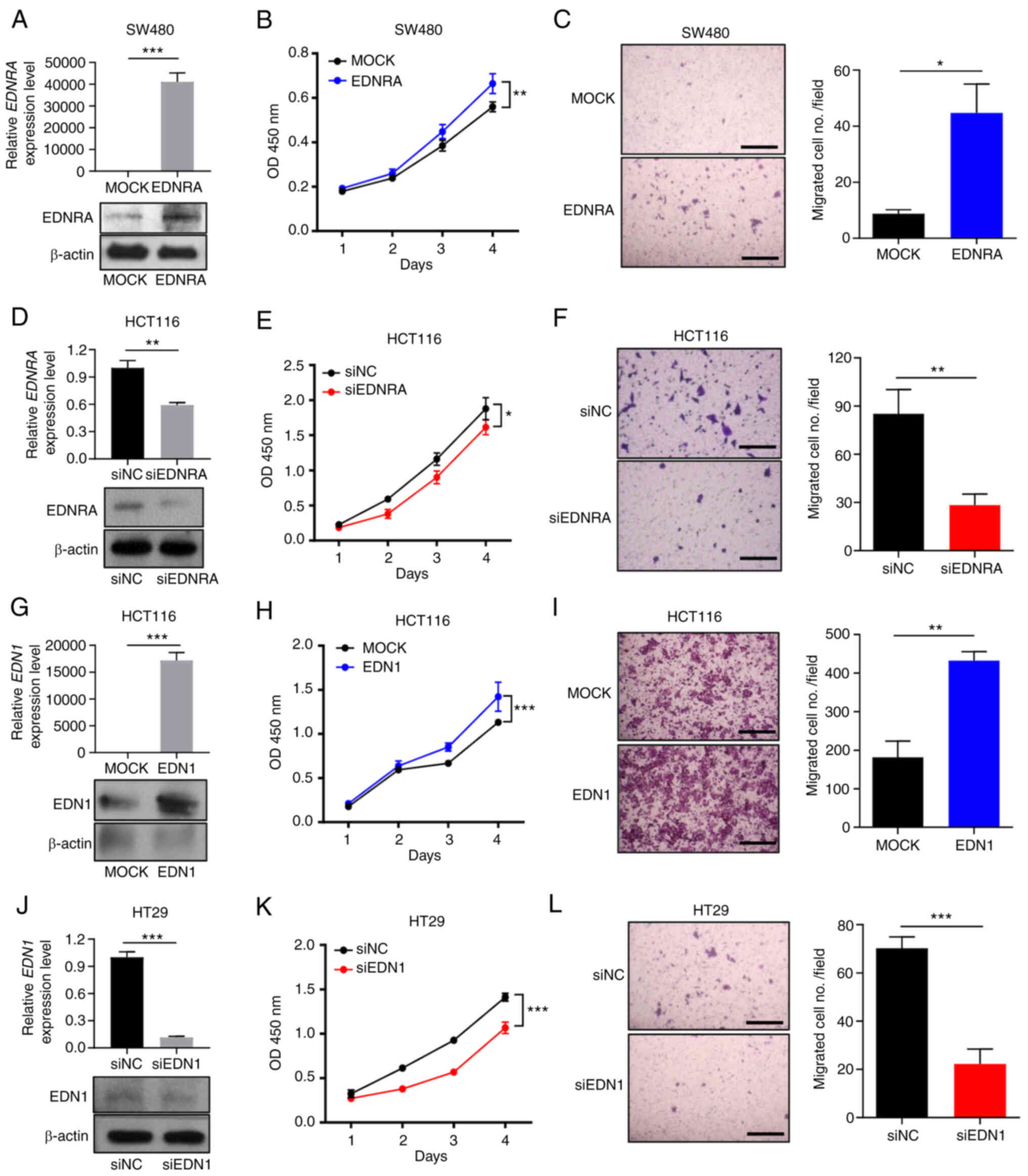 | Figure 2Expression levels of EDN1 and EDNRA
regulate the growth and migratory ability of colorectal cancer
cells. (A) EDNRA-overexpressing SW480 cells were constructed, and
EDNRA expression levels were measured at the mRNA level and protein
level using RT-qPCR and western blotting, respectively. EDNRA
overexpression increased (B) proliferation and (C) migration
compared to mock transfection in SW480 cells. Cell proliferation
was measured using CCK-8 assay. Transwell membranes were fixed and
stained with crystal violet. The presented images indicate stained
migrating cells (scale bar, 500 µm) and the number of
relative migrated cells. (D) HCT116 cells transfected with siEDNRA
demonstrated decreased EDNRA expression levels compared with
siNC-transfected cells. Knockdown of EDNRA decreased (E)
proliferation and (F) migration compared to the negative control in
HCT116 cells. Cell proliferation was measured by using the CCK-8
assay. Transwell membranes were fixed and stained with crystal
violet. The presented images indicate stained migrating cells
(scale bar, 500 µm) and the number of relative migrated
cells. (G) EDN1-overexpressing HCT116 cells were constructed, and
EDN1 expression levels were determined at the mRNA and protein
levels using RT-qPCR and western blotting, respectively. The
overexpression of EDN1 increased (H) proliferation and (I)
migration compared to mock transfection of HCT116 cells. Cell
proliferation was measured using CCK-8 assay. Transwell membranes
were fixed and stained with crystal violet. The presented images
indicate stained migrating cells (scale bar, 500 µm) and the
number of relative migrated cells. (J) HT29 cells transfected with
siEDN1 displayed decreased EDN1 expression levels compared with
siNC-transfected cells. The knockdown of EDN1 decreased (K)
proliferation and (L) migration compared to the negative control in
HT29 cells. Cell proliferation was measured using CCK-8 assay.
Transwell membranes were fixed and stained with crystal violet. The
presented images indicate stained migrating cells (scale bar, 500
µm) and the number of relative migrated cells. Data were
analyzed using a Student's t-test; *P<0.05,
**P<0.01 and ***P<0.001. EDN1,
endothelin-1; EDNRA, endothelin receptor A; siNC, negative control
siRNA; CCK-8, Cell Counting Kit-8; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Furthermore, in order to ascertain the cellular
functions of EDN1, firstly we analyzed the mRNA expression level of
EDN1 in six different CRC cell lines (Fig. S4A). The EDN1-overexpression vector
was transfected into HCT116 cells (Fig. 2G), and cell proliferation and
migration assays were performed. Subsequently, the growth of HCT116
cells significantly increased (Fig.
2H). In addition, a significantly increased number of migrated
cells was observed in the EDN1 overexpression group (Fig. 2I). By contrast, the knockdown of
EDN1 (Figs. 2J and S4B) significantly inhibited the
proliferation and motility of HT29 and SW620 cells (Figs. 2K and L, and S4C and D). Thus, these findings
demonstrated that EDN1 and EDNRA may play a crucial role in CRC
progression.
Knockdown of EDN1 and EDNRA induces the
apoptosis of CRC cells
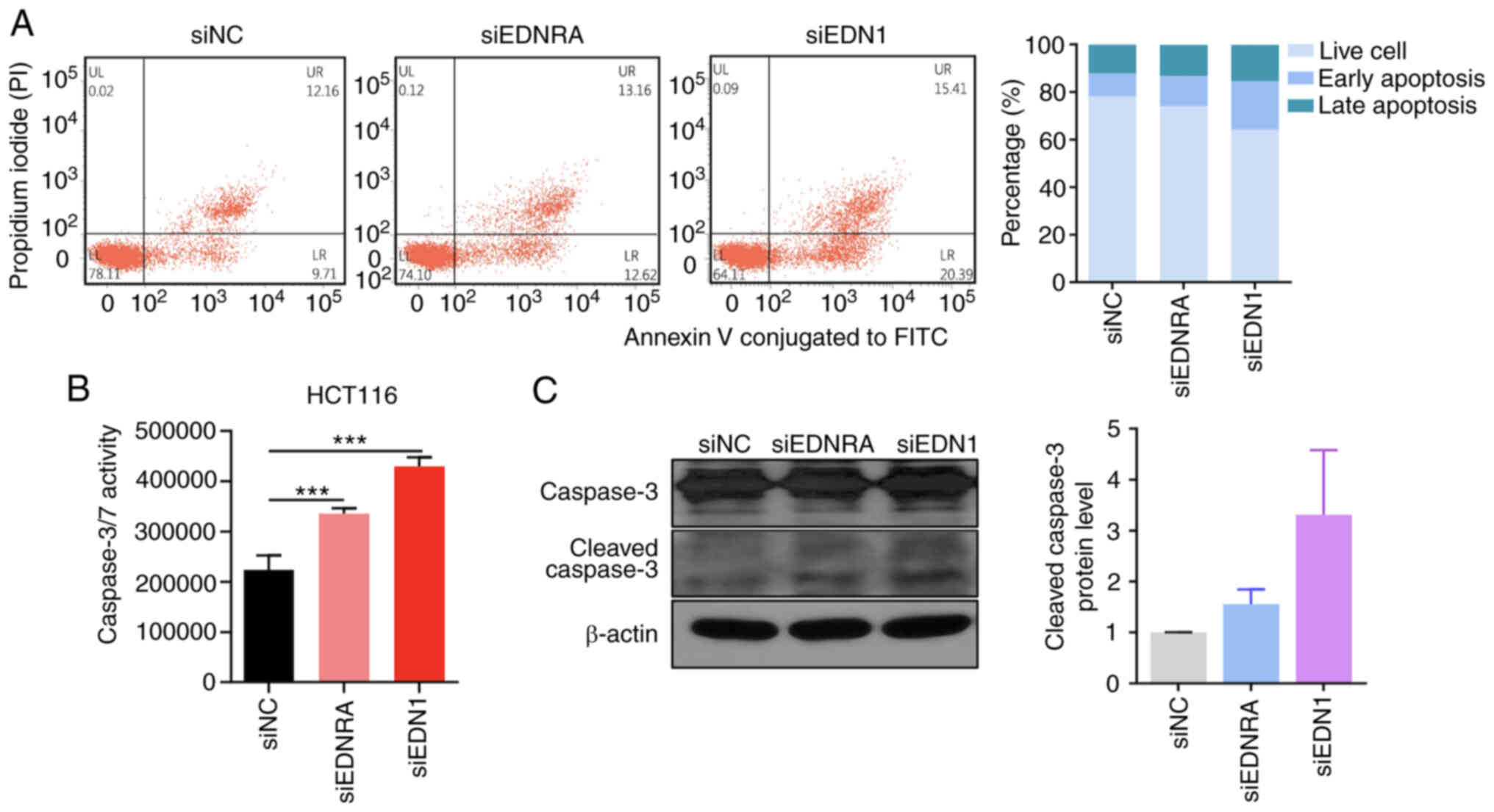
In order to demonstrate the mechanisms through which
EDNRA and EDN1 regulate the growth of CRC cells, flow cytometric
analysis was performed, following the transfection of HCT116 cells
with siRNA. Annexin V and PI double staining was used to detect
apoptotic cells, and the ratio of early and late apoptotic cells
was quantified. As depicted in Fig.
3A, the apoptotic cell numbers were increased in the siEDNRA-
and siEDN1-transfected cells, as compared to the siNC-transfected
cells. Similarly, the caspase-3/7 assay revealed that the knockdown
of EDNRA or EDN1 significantly increased caspase-3/7 activity in
the HCT116 cells (Fig. 3B). In
addition, the transfection of the HCT116 cells with siEDNRA or
siEDN1 increased the cleaved caspase-3 form (Fig. 3C). Overall, the inactivation of the
EDN1/EDNRA axis suppressed cell growth by inducing cellular
apoptosis.
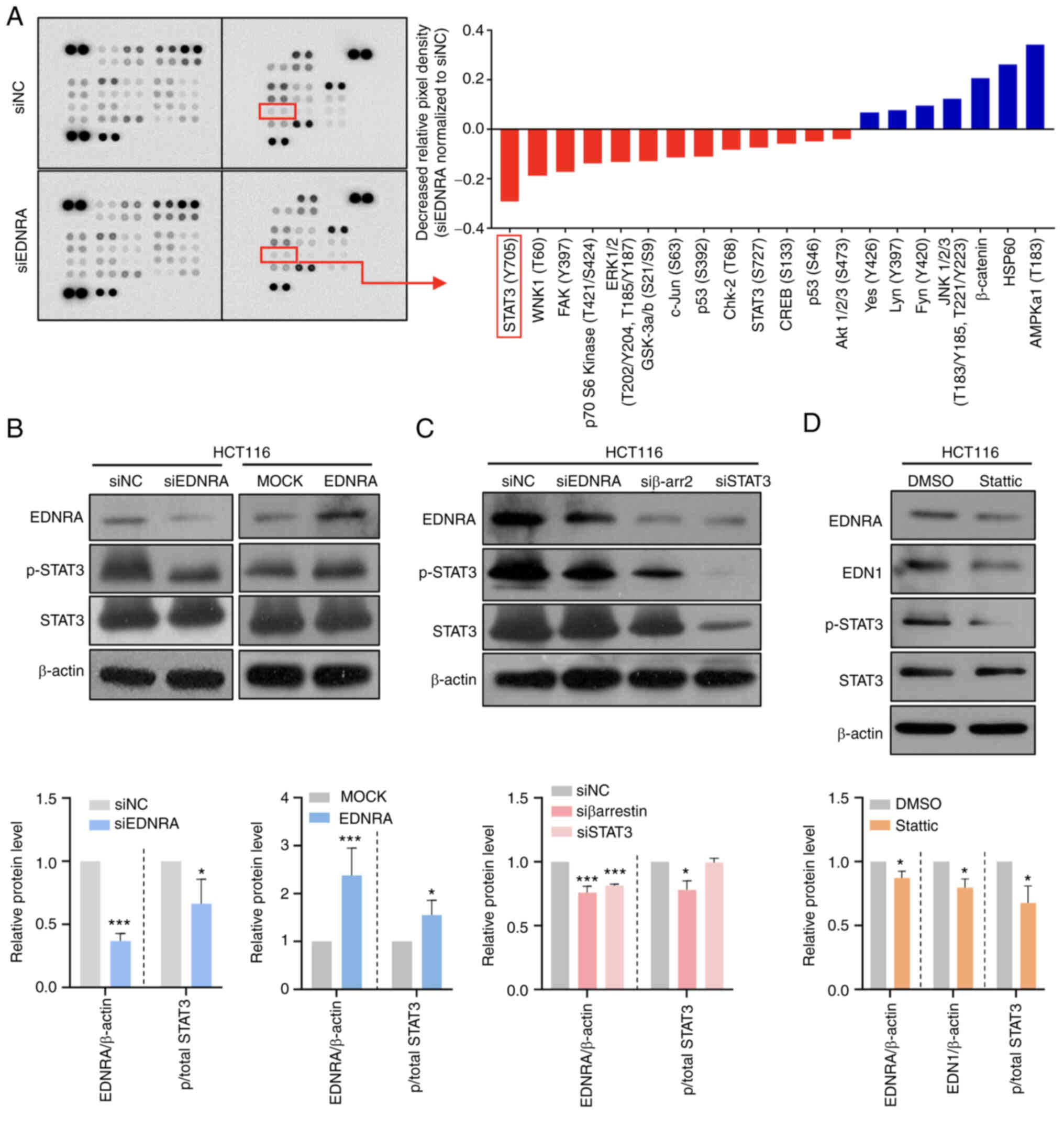
EDNRA modulates the STAT3 signaling
pathway in CRC cells
In order to further elucidate the molecular
mechanisms underlying EDNRA-induced oncogenic functions, a
phosphokinase array assay in HCT116 cells transfected with siNC or
siEDNRA was performed. Of note, decreased levels of p-STAT3 (Y705)
and p-p70 S6 kinase (T389) in siED- NRA-transfected cells, as
compared to siNC-transfected cells were observed (Fig. 4A). To verify the array data,
western blot analysis was conducted, using HCT116 cells transfected
with siNC or siEDNRA. No significant difference in the
phosphorylation of p70S6K between the siNC and siEDNRA groups
(Fig. S5A) was observed; however,
the p-STAT3 (Y705) level was significantly reduced in
siEDNRA-transfected HCT116 cells, while the p-STAT3 levels were
highly increased in the EDNRA-overexpressing HCT116 cells (Fig. 4B).
Subsequently, a knockdown experiment of the
downstream activator, β-arrestin, was performed in the HCT116 cells
to determine the mechanisms through which the p-STAT3 level is
regulated by the EDN1/EDNRA axis. For the analysis of these, the
EDRNA, p-STAT3 and STAT3 protein levels were evaluated using
western blot analysis. The EDNRA protein level was significantly
decreased in the siβ-arrestin2 (siβ-arr2)- or siSTAT3-transfected
cells (Fig. 4C), suggesting that
EDN1-EDNRA-β-arrestin-STAT3 signal transduction may present a
positive feedback loop. In addition, the supporting evidence of the
positive feedback loop from the COAD TCGA dataset resulted in a
significant positive correlation of transcriptional levels between
genes EDN1, EDNRA, ARRB1, ARRB2 and STAT3 (Fig. S5B and C).
To confirm that EDN1 and EDNRA are regulated by
STAT3, the HCT116 cells were treated with the STAT3 inhibitor,
static (32), which
dephosphorylates Y705 and S727 of STAT3. Notably, the protein
levels of both EDN1 and EDNRA were significantly decreased by the
STAT3 inhibitor (Fig. 4D),
indicating that EDN1 and EDNRA expression can be modulated by
regulating STAT3 phosphorylation.
Induction of EDN1 and EDNRA expression
via the STAT3 pathway
The suppression of STAT3 expression using siRNA
significantly and concurrently decreased the EDN1 and
EDNRA mRNA expression levels in HCT116 cells (Fig. 5A). To further elucidate the
mechanisms through which STAT3 regulates EDN1 and EDNRA, the EPD
database (https://epd.epfl.ch/EPDnew_database.php) was queried
concerning this issue, revealing that STAT3 bound to the EDN1 or
EDNRA proximal promoters. In Fig.
5B, the STAT3 consensus binding site with position frequency
matrix (Matrix ID: MA0144.2) sequence logo was illustrated from the
JASPAR database version 2022 (http://jaspar.genereg.net/analysis). Furthermore, a
ChIP assay was performed in the predicted promoter region, using
HCT116 cells transfected with siNC or siSTAT3. The ChIP-qPCR
results revealed that siSTAT3 significantly decreased the fold
enrichment for EDN1 or EDNRA expression compared to siNC (Fig. 5C and D), indicating that STAT3
directly binds to the promoter regions of EDN1 and EDNRA, to
regulate their expression.
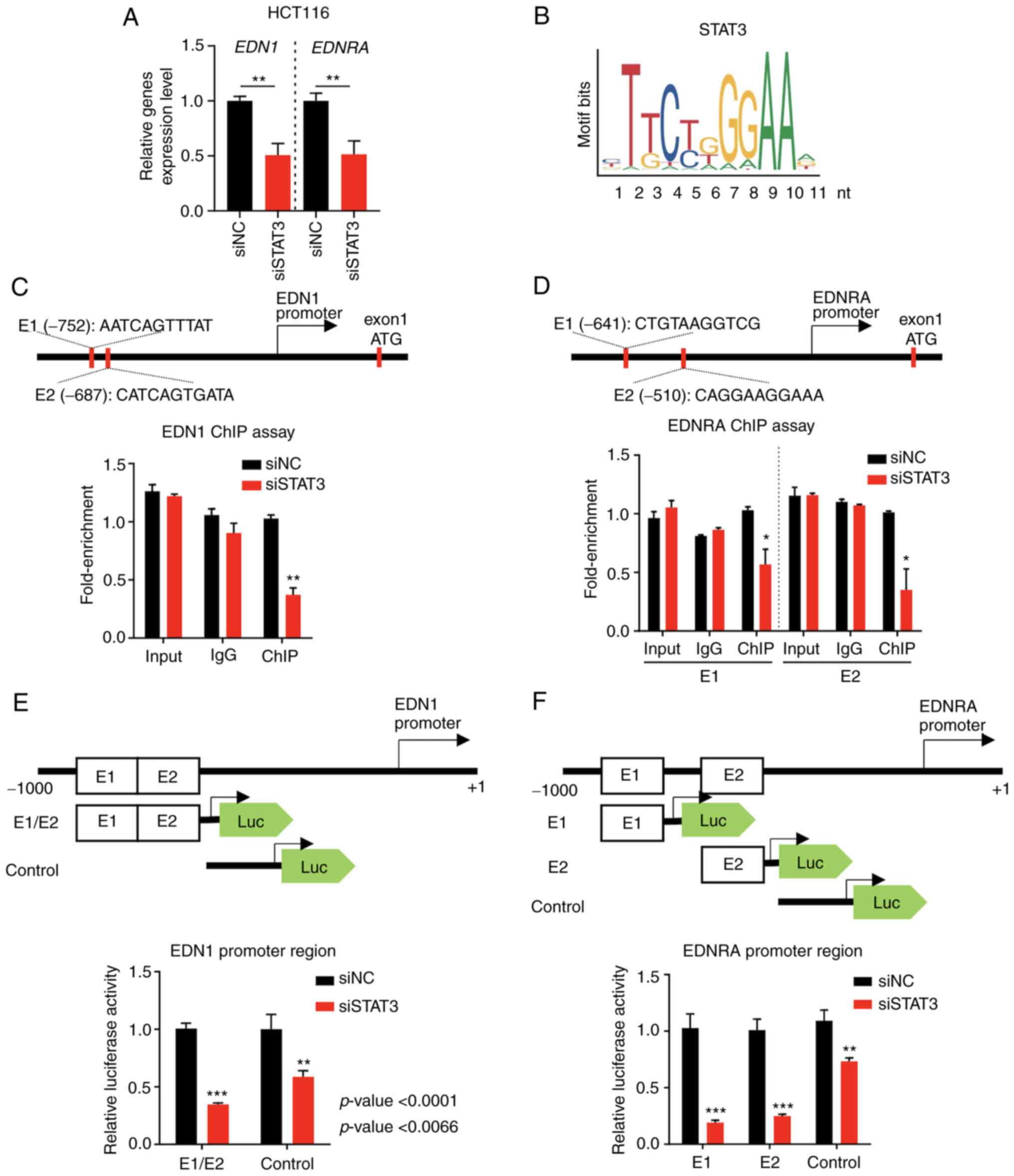 | Figure 5The transcription of EDN1 and
EDNRA is regulated by the activity of the transcription
factor STAT3. (A) The mRNA levels of EDN1 and EDNRA
were decreased in HCT116 cell lines transfected with siSTAT3
compared to siNC. (B) The binding motif of STAT3 for the regulation
of the transcription of target genes. (C) The putative
STAT3-binding motif of EDN1 was predicted under the condition of a
P=0.001, and the ChIP assay confirmed the regulation of EDN1
transcription by the transcription factor STAT3. Data were analyzed
using a Student's t-test. (D) The putative STAT3-binding motif of
EDNRA was predicted (P<0.0001), and the ChIP assay confirmed the
regulation of EDNRA transcription by the transcription
factor STAT3. Data were analyzed using a Student's t-test. (E) The
EDN1 promoter region containing the STAT3-binding site was
cloned into the pGL4 vector, and luciferase activity was measured.
Data were analyzed using a Student's t-test. (F) The EDNRA
promoter region containing the STAT3-binding site was cloned into
the pGL4 vector, and luciferase activity was measured. The results
confirmed that EDN1 and EDNRA promoter activity was
reduced in siSTAT3 HCT116 cells. Data were analyzed using a
Student's t-test; *P<0.05, **P<0.01 and
***P<0.001. EDN1, endothelin-1; EDNRA, endothelin
receptor A; siNC, negative control siRNA; ChIP, chromatin
immunoprecipitation; STAT3, signal transducer and activator of
transcription 3. |
The role of STAT3 in EDN1 and EDNRA
mRNA transcription by constructing luciferase reporter vectors was
then examined using the EDN1 or EDNRA gene promoter that contained
each pair of putative STAT3-binding sites. The co-transfection of
siNC or siSTAT3 with the luciferase reporter vector indicated that
the siRNA-mediated suppression of STAT3 decreased the luciferase
activity of both the EDN1 and EDNRA constructs (Fig. 5E and F). Taken together, ChIP and
promoter assays strongly indicated that STAT3 may directly regulate
EDN1 and EDNRA expression at the transcriptional
level, by binding to their promoter regions.
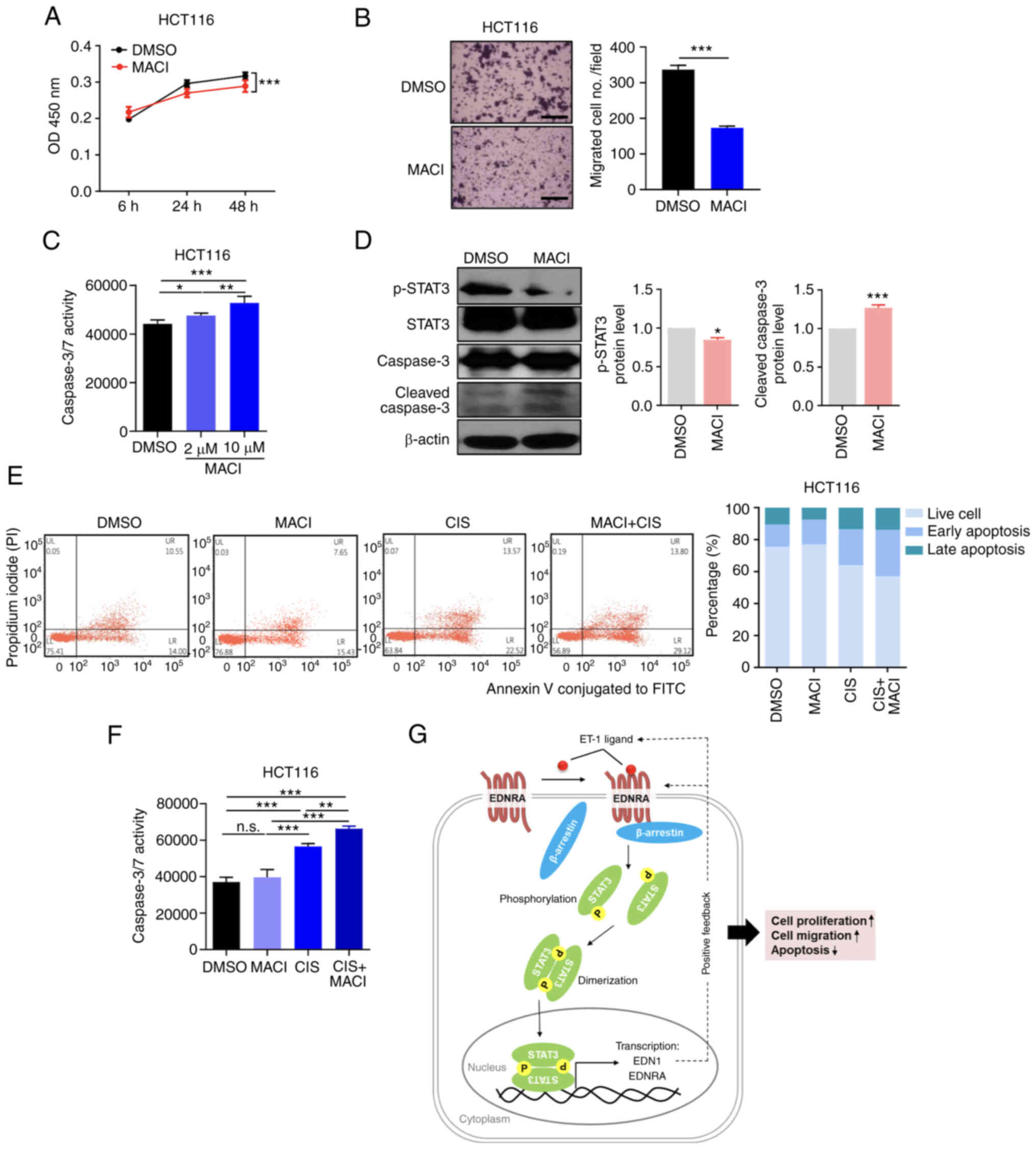
Pharmacological inhibition of EDNRA
Macitentan, known as a dual endothelin receptor
antagonist, is a Food and Drug Administration (FDA)-approved drug
that is being used as a treatment for pulmonary hypertension
disease (33-35). Based on the role of EDNRA in the
progression of CRC, the possibility that macitentan, which was
approved as a drug for pulmonary hypertension by the US FDA in
2013, could be repurposed as an anticancer drug for CRC, was
investigated herein. Treatment of the HCT116 cells with macitentan
slightly suppressed cell growth and significantly reduced cell
motility (Fig. 6A and B).
Furthermore, it was revealed that treatment of the HCT116 cells
with macitentan led to a significant increase in the number of
apoptotic cells, according to the results of a Caspase-Glo 3/7
assay (Fig. 6C). Furthermore,
western blot analysis demonstrated that macitentan reduced the
p-STAT3 levels and increased the cleaved caspase-3 levels (Fig. 6D).
To further assess the combined therapeutic effects
of macitentan and cisplatin on CRC cells, an apoptosis assay was
performed. FACS analysis demonstrated that combination therapy with
cisplatin and macitentan increased the apoptotic cell numbers, in
comparison to treatment with macitentan or cisplatin alone
(Figs. 6E and S6A). Similarly, apoptosis analysis using
the Caspase-Glo 3/7 assay revealed that the apoptotic cell numbers
were increased in cells treated with macitentan or cisplatin and
even further in cells treated with the combination treatment
(Figs. 6F and S6B). Collectively, the aforementioned
data suggest that the pharmacological EDN1/EDNRA blockade by
macitentan, in combination with chemotherapeutic agents, may be a
therapeutic option for patients with CRC, by inducing the apoptosis
of CRC cells and suppressing STAT3 signaling.
Discussion
GPCRs are membrane receptor proteins that play a
crucial role in various cellular processes by transmitting various
extracellular signals, triggered by specific peptides, ions,
hormones and photons, into cells, in order to regulate cell
proliferation, differentiation, and communication. Emerging
evidence indicates that abnormally expressed GPCRs are involved in
various diseases, including cancers (36). Therefore, blocking GPCRs and their
downstream target molecules may be a promising treatment strategy.
Moreover, almost 35% of marketed drugs that have been approved by
the FDA target GPCRs (37). EDNRA
is a well-known GPCR, that regulates blood pressure and acts as a
vasoconstrictor when bound to its ligand, EDN1. However, the
aberrant expression and activation of EDN1 and EDNRA have been
reported to be associated with a number of diseases, including
hypertension, diabetes, obesity and cancers. For instance, the
importance of EDNRA has been reported in the chemotherapeutic
resistance of ovarian cancer, resulting in a high expression of
EDNRA related to poor clinical outcomes and chemoresistance by
activating the Wnt/β-catenin pathway (38). Consistent with previous reports,
demonstrating the overexpression of EDRNA in prostate, colon,
breast and cervical cancers (21,27),
the present study also observed that EDNRA expression was
upregulated in CRC, and the survival rate of patients with CRC with
a high expression of EDNRA was decreased. A positive correlation
between the EDN1 and EDNRA levels was detected in the
COAD TCGA data set. In addition, the in vitro experimental
results of the present study revealed that silencing EDN1 or EDNRA
suppressed cell growth and migratory abilities, suggesting that the
suppression of EDN1 and EDNRA may effectively attenuate oncogenic
properties; thus, EDNRA may be an important therapeutic target for
blocking CRC progression.
Although the HCT116, SW620 and KM12SM cell lines
that have been reported to have high invasiveness or metastatic
potential displayed moderate to high expression levels of EDNRA
in vitro, it could not be concluded that EDNRA expression
was strongly associated with CRC progression, as there was no
particular association pattern between disease stage and EDNRA
expression level in vivo, according to the COAD TCGA data
set analysis. Animal experiments with the orthotopic implantation
of a pair of parental and engineered cells manipulated to
overexpress or knockout EDN1/EDNRA will aid to delineate the
inconsistency between in vitro and in vivo data and,
clearly elucidating the role of EDN1/EDNRA in the progression of
CRC.
EDNRB functions have been reported as a
counter-regulator of EDN1/EDNRA activity via various mechanisms,
including nitric oxide production, EDN1 clearance and blocking cell
growth in normal cells (39,40).
Compared to EDRNA, the detailed mechanisms and functions of EDNRB
have not yet been completely elucidated in cancer research, since
the effects of EDN1 on cancer cells are mostly mediated by EDNRA.
Although a positive correlation between EDN1 and EDNRB expression
was observed (Fig. S1D), the
findings that both EDN1 and EDNRB expression levels were
significantly lower in CRC tissues, as compared with normal tissues
do not support the clinical significance of the correlation data.
It has been previously reviewed that EDNRB expression was
upregulated in bladder cancer and was associated with a poor
prognosis (41). In addition, Fu
et al (42) reported that
miR-124-3p suppressed cell proliferation and invasion by the direct
regulation of EDNRB in bladder cancer. By contrast, EDNRB
expression has been shown to be downregulated by aberrant
hypermethylation in the promoter site in several tumors, including
hepatocellular carcinoma, gastric cancer and CRC (43-45).
A reduced EDNRB expression by hypermethylation has been found to be
associated with gastric cancer invasion and to be associated with
pathological stage in prostate cancer (46). Similarly, the results of the
present study also revealed that the expression of EDNRA was more
prominent than EDNRB in CRC, indicating that EDNRA may play a
critical role in CRC progression.
Although EDNRA expression was upregulated in CRC
tumors and had a positive correlation with EDN1 ligand, EDN1
expression was lower in CRC tumors than in normal tissues. This may
raise a question whether other ligands exist which are able to bind
and activate EDNRA-mediated signaling. It is possible that other
ligands, such as EDN2 can also bind to EDNRA with different
affinities than EDN1 and can affect EDNRA signaling independently
of EDN1 ligand in CRC. In addition, EDN1 could also be derived from
cells in the tumor microenvironment including immune cells,
fibroblasts and blood vessels and may be used to activate EDNRA
signaling in a paracrine manner (47,48).
In this regard, single-cell transcriptomic analysis of CRC tumor
tissues in the future is expected to provide further information
about the role of autocrine and/or paracrine EDN1 in the
EDNRA-mediated growth and progression of CRC.
Accumulating evidence has indicated that EDNRA plays
a crucial role in carcinogenesis by regulating several downstream
signaling pathways. For example, Cianfrocca et al (49) revealed a mechanistic link between
EDN1/EDNRA signaling and the β-catenin pathway through a specific
interaction with β-arrestin in CRC stem-like cells. Another study
demonstrated that EDNRA was directly regulated by miR-200c in
gastric cancer (50). The EDNRA
3′-untranslated region encodes a binding site of miR-200c;
therefore, EDNRA can be regulated by miRNAs. Notably, Sestito et
al (51) observed that EDNRA
and ZEB1 negatively correlated with miR-200b/c, and the
EDNRA/miR-200b/c/ZEB1 circuit promoted epithelial-mesenchymal
transition, cell plasticity and metastasis. These results indicated
that EDNRA and its associated signaling pathway may be important
for tumorigenesis. In the present study, it was also investigated
how EDN1/EDNRA may serve its oncogenic role by surveying putative
downstream effector molecules that can enhance CRC progression. The
phosphokinase array demonstrated that EDNRA silencing decreased the
phosphorylation of STAT3 in CRC cells, indicating that EDN1/EDNRA
may be involved in STAT3 activation. Therefore, it was hypothesized
that EDNRA may promote CRC progression through the activation of
STAT3 signaling pathways. STAT3 is a member of the STAT family of
transcription factors, and the JAK-STAT signaling pathway is a
well-established mechanism that regulates various cell functions,
including immunity, cell division and tumorigenesis, through the
regulation of the transcription level of its target genes (52). Presenting with these multimodal
tumor promoting-activities, it is unsurprising that p-STAT3 levels
are increased in CRC (53,54), and its activation has been shown to
be associated with cancer progression (55). In the present study, a novel
mechanism, to the best of our knowledge, of the EDN1/EDNRA-mediated
CRC progression was revealed in that EDNRA expression can promote
STAT3 phosphorylation; thus, STAT3-driven tumor-promoting
activities may be activated in CRC cells. These oncogenic
properties can be further supported by a positive feedback
regulatory circuit between STAT3 activation and transcriptional
upregulation of EDN1/EDNRA expression. Therefore, the present study
provided a strong missing link between STAT3 activation and
EDN1/EDNRA for the coordination of tumor progression in CRC.
Additionally, the present study is the first to
demonstrate, to the best of our knowledge, that STAT3
simultaneously regulates EDN1 and EDNRA mRNA expression. The
inactivation of STAT3 by siRNA or STAT3 inhibitor stattic resulted
in the concurrent suppression of EDN1 and EDNRA expression levels,
indicating that STAT3 may regulate the expression of EDN1 and
EDNRA. By using ChIP-qPCR and promoter assays, it was confirmed
that EDN1 and EDNRA were directly regulated by the STAT3
transcription factor. EDN1 has been reported to activate the
EDNRA/β-arrestin/STAT3 loop, and this circuit most likely can also
be induced by other extrinsic or CRC cell intrinsic signaling
pathways, such as the activation of oncogenes and inactivation of
tumor suppressors (56,57), that may activate EDN1, EDNRA or
STAT3. Once activated, the circuit can be continuously active
through its positive feed-forward nature, as many tumor-promoting
oncogenes do. Overall, it was suggested that the EDN1/EDNRA
signaling pathway promotes various downstream effector proteins,
including STAT3; subsequently, these effector proteins may
positively regulate the transcriptional levels of EDN1 or EDNRA. To
the best of our knowledge, this is the first report demonstrating
STAT3-mediated overexpression of EDN1 and EDNRA in CRC and its
functional significance. However, the present study has a
limitation concerning a lack of a full explanation about how the
EDN1/EDNRA axis may be associated with STAT3 activation, since
STAT3 can also be activated by several upstream signals, including
the inflammation-related receptors gp130, IL-6R, IL-8R, IL-35R and
EGFR; thus, further studies are required for the determination of
the sophisticated regulatory networks between EDNRA expression and
other signaling pathways, including inflammatory signaling in CRC.
Moreover, it remains to be fully elucidated whether the exact role
of EDN1/EDNRA activation and its downstream STAT3 signaling favor
tumor-initiation or tumor-promotion. To delineate this, animal
experiments using genetically engineered mice with EDN1 or EDNRA
overexpression in the normal intestinal cells need to be performed
in future studies, in order to determine whether non-cancerous or
pre-cancerous cells may transform into cancer cells.
In addition, other signaling pathways, including
with-no-lysine [K] 1 (WNK1) and/or focal adhesion kinase (FAK)
signaling can also contribute to the EDRA-mediated progression of
CRC, although no experimental evidence was presented in the present
study, since the phosphokinase array data analysis of the present
study demonstrated decreased phosphorylation levels of WNK1 and/or
FAK by EDNRA silencing. WNK kinases have been reported to activate
downstream substrates, including protein odd-skipped-related 1 and
STE20/SPS1-related proline-alanine-rich protein kinase to regulate
several ion channels and to stimulate angiogenesis by stimulating
PI3K-AKT pathway (58). Moreover,
WNK kinase regulates Wnt signaling pathways and β-Catenin levels,
which are both associated with the development and progression of
CRC (59). By contrast, FAK is a
well-known multi-functional regulator of cell signaling within the
tumor microenvironment (60). It
promotes cell proliferation, motility and survival through a
kinase-dependent or -independent manner in several cancers,
including colon, breast, ovary and prostate cancers (61). Integrin α5 has been reported to
promote cell migration and invasion through the activation of
FAK/STAT3/AKT signaling pathways (62). In addition to EDNRA, FAK can also
activate Src/ERK1/2/STAT3 signaling to inhibit
E-cadherin expression, thus leading to the promotion of cell
motility in melanoma cells (63).
Currently, it is not clear whether the activation of STAT3 by EDNRA
is FAK-dependent. Moreover, the exact mechanism about the
interactions between all signaling molecules that are involved in
the EDNRA-mediated CRC progression remains to be fully elucidated.
Accordingly, to fully understand the EDNRA-mediated signaling
networks and to evaluate the biochemical and clinical significance
of the current findings, further experimental evidence is required
in the future to delineate biochemical roles of those signaling
molecules on the progression of CRC tumors.
Furthermore, considering the findings of the present
study in light of recent studies demonstrating that the endothelin
receptor antagonist, macitentan, may be used as a therapeutic drug
for targeting various cancer types apart from pulmonary
hypertension disease (64).
Accordingly, previous reports have revealed that targeting
endothelin receptors with macitentan represent an effective
therapeutic approach for anti-proliferation and anti-angiogenesis
in multiple myeloma and high-grade serous ovarian cancer (65,66).
Notably, a combination therapeutic strategy with macitentan and
chemotherapeutic agents, including oxaliplatin and 5-fluorouracil
has been indicated to exert enhanced antitumor effects in CRC
(49). Lee et al (67) revealed a reduction in cell division
and induction of apoptosis by macitentan combined with paclitaxel
in breast and lung cancer cells, also significantly increasing
overall survival in mice harboring brain metastases. Another group
reported that macitentan in combination with temozolomide also
leads to glioblastoma regression (68). Similarly, the results of the
present study also demonstrated that macitentan effectively
inhibited cell growth and migration and induced apoptosis of CRC
cells, particularly when used in combination with the
chemotherapeutic agent cisplatin. More specifically, combined
treatment of macitentan and cisplatin induced tumor cell apoptosis
more effectively than treated alone, possibly by blocking different
apoptotic signaling pathways, since the mechanism of apoptosis
induced by macitentan appears to be mainly mediated by blocking
MAPK and/or STAT signaling pathways, whereas cisplatin induces p53
signaling pathway through proapoptotic FasL gene expression
(69,70). The clinical significance of the
present findings need to be evaluated in the clinical setting, and
the data suggested that the EDN1/EDNRA blockade may be possibly
used as a as a novel treatment option for patients with CRC.
Although cisplatin is a highly effective anticancer
drug widely used for the treatment of several types of cancer in
the clinic (71,72), standard chemotherapy including
leucovorin, irinotecan and 5-fluorouracil (73,74)
but not cisplatin has been used for the treatment of patients with
CRC. Therefore, those chemotherapeutics should have been used
rather than cisplatin in the present experimental setup to confirm
the anticancer effects of combination therapy with EDNRA antagonist
and chemotherapeutic agents against CRC. Regardless of the above
limitation, the findings of the present study may provide
preliminary data presenting the possibility for the EDNRA
antagonists being able to improve chemotherapy outcomes, when
treated in combination. Additionally, it will be interesting to
perform a combination treatment of macitentan with stattic to
evaluate whether the blockade of STAT3 and/or EDNRA signaling,
alone or in combination, can improve the therapy outcomes for the
therapy of patients with CRC. Therefore, further in vivo
studies to evaluate the therapeutic efficacy of various EDNRA
antagonists, and/or STAT3 signaling antagonists, in combination
with variety of chemotherapeutic agents for the therapy of patients
with CRC are warranted.
In the present study, it was demonstrated that
EDN1/EDNRA, being highly expressed in human CRC, promoted CRC cell
proliferation and migration, and decreased apoptosis through the
β-arrestin-mediated activation of the well-known oncogenic protein
STAT3 (Fig. 6G). The activation of
STAT3, in turn, continuously activated EDN1/EDNRA signaling by
increasing the transcription of several genes associated with tumor
progression, including EDN1/EDNRA, in a positive
feedback loop. Based on these findings, considering all data
demonstrating that the pharmacological inhibition of EDNRA
signaling, in combination with chemotherapy, inhibits the
proliferation and migration and induces apoptosis of CRC cells, it
was proposed that targeting EDN1/EDNRA signaling may be an
effective therapeutic strategy against CRC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
YJL, EJ, JC, TSH and JSK designed the experiments.
YJL, EJ, JC, JSH, EJJ and YNR performed the experiments and
collected the data. HSB, SK, SKK and SYK analyzed the
bioinformatics data. EJ, HSB, JKM, TSH and JSK analyzed and
interpreted the data. YJL, EJ, JC, TSH and JSK drafted the
manuscript and revised the manuscript. TSH and JSK confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
EDNRA
|
endothelin receptor A
|
|
EDN1
|
endothelin-1
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
CRC
|
colorectal cancer
|
|
GPCR
|
G protein-coupled receptor
|
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the National
Research Foundation of Korea (NRF) grant funded by the Korean
Government (Ministry of Science and ICT, MSIT; grant no.
NRF-2020R1C1C1007431), the Korean Fund for Regenerative Medicine
(KFRM) grant funded by the Korea government (the Ministry of
Science and ICT, the Ministry of Health and Welfare; grant no.
21A0404L1), and the KRIBB Research Initiative Program (grant no.
1711170580).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Sauer AG, Fedewa SA,
Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal A:
Colorectal cancer statistics, 2020. CA Cancer J Clin. 70:145–164.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Murphy CC, Harlan LC, Lund JL, Lynch CF
and Geiger AM: Patterns of colorectal cancer care in the United
States: 1990-2010. J Natl Cancer Inst. 107:djv1982015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Keum N and Giovannucci E: Global burden of
colorectal cancer: emerging trends, risk factors and prevention
strategies. Nat Rev Gastroenterol Hepatol. 16:713–732. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanchez-Gundin J, Fernandez-Carballido A
M, Martinez-Valdivieso L, Barreda-Hernandez D and Torres-Suarez AI:
New trends in the therapeutic approach to metastatic colorectal
cancer. Int J Med Sci. 15:659–665. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oh DY and Bang YJ: HER2-targeted
therapies-a role beyond breast cancer. Nat Rev Clin Oncol.
17:33–48. 2020. View Article : Google Scholar
|
|
7
|
Ferguson FM and Gray NS: Kinase
inhibitors: The road ahead. Nat Rev Drug Discov. 17:353–377. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meric-Bernstam F and Mills GB: Overcoming
implementation challenges of personalized cancer therapy. Nat Rev
Clin Oncol. 9:542–548. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sala E, Mologni L, Truffa S, Gaetano C,
Bollag GE and Gambacorti-Passerini C: BRAF silencing by short
hairpin RNA or chemical blockade by PLX4032 leads to different
responses in melanoma and thyroid carcinoma cells. Mol Cancer Res.
6:751–759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chung CH, Mirakhur B, Chan E, Le QT,
Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, et
al: Cetuximab-induced anaphylaxis and IgE specific for
galactose-alpha-1,3-galactose. N Engl J Med. 358:1109–1117. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cohen MH, Gootenberg J, Keegan P and
Pazdur R: FDA drug approval summary: Bevacizumab plus FOLFOX4 as
second-line treatment of colorectal cancer. Oncologist. 12:356–361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Le XF, Pruefer F and Bast RC Jr:
HER2-targeting antibodies modulate the cyclin-dependent kinase
inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle.
4:87–95. 2005. View Article : Google Scholar
|
|
13
|
Curigliano G and Criscitiello C: Successes
and limitations of targeted cancer therapy in breast cancer. Prog
Tumor Res. 41:15–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Holcmann M and Sibilia M: Mechanisms
underlying skin disorders induced by EGFR inhibitors. Mol Cell
Oncol. 2:e10049692015. View Article : Google Scholar
|
|
15
|
Welsh SJ and Corrie PG: Management of BRAF
and MEK inhibitor toxicities in patients with metastatic melanoma.
Ther Adv Med Oncol. 7:122–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spaans JN and Goss GD: Drug resistance to
molecular targeted therapy and its consequences for treatment
decisions in non-small-cell lung cancer. Front Oncol. 4:1902014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yanagisawa M, Kurihara H, Kimura S, Tomobe
Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K and Masaki T: A novel
potent vasoconstrictor peptide produced by vascular endothelial
cells. Nature. 332:411–415. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Inoue A, Yanagisawa M, Kimura S, Kasuya Y,
Miyauchi T, Goto K and Masaki T: The human endothelin family: Three
structurally and pharmacologically distinct isopeptides predicted
by three separate genes. Proc Natl Acad Sci USA. 86:2863–2867.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davenport AP, Hyndman KA, Dhaun N, Southan
C, Kohan DE, Pollock JS, Pollock DM, Webb DJ and Maguire JJ:
Endothelin. Pharmacol Rev. 68:357–418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakurai T, Yanagisawa M, Takuwa Y,
Miyazaki H, Kimura S, Goto K and Masaki T: Cloning of a cDNA
encoding a non-isopeptide-selective subtype of the endothelin
receptor. Nature. 348:732–735. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosanò L, Spinella F and Bagnato A:
Endothelin 1 in cancer: Biological implications and therapeutic
opportunities. Nat Rev Cancer. 13:637–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bagnato A and Natali PG: Endothelin
receptors as novel targets in tumor therapy. J Transl Med.
2:162004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosanò L and Bagnato A: Endothelin
therapeutics in cancer: Where are we? Am J Physiol Regul Integr
Comp Physiol. 310:R469–R475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bondurand N, Dufour S and Pingault V: News
from the endothelin-3/EDNRB signaling pathway: Role during enteric
nervous system development and involvement in neural
crest-associated disorders. Dev Biol. 444(Suppl 1): S156–S169.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kohan DE, Rossi NF, Inscho EW and Pollock
DM: Regulation of blood pressure and salt homeostasis by
endothelin. Physiol Rev. 91:1–77. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nelson JB, Udan MS, Guruli G and Pflug BR:
Endothelin-1 inhibits apoptosis in prostate cancer. Neoplasia.
7:631–637. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Smith MP, Rowling EJ, Miskolczi Z,
Ferguson J, Spoerri L, Haass NK, Sloss O, McEntegart S, Arozarena
I, von Kriegsheim A, et al: Targeting endothelin receptor
signalling overcomes heterogeneity driven therapy failure. EMBO Mol
Med. 9:1011–1029. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laurberg JR, Jensen JB, Schepeler T, Borre
M, Orntoft TF and Dyrskjot L: High expression of GEM and EDNRA is
associated with metastasis and poor outcome in patients with
advanced bladder cancer. BMC Cancer. 14:6382014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Minchenko DO, Tsymbal DO, Riabovol OO,
Viletska YM, Lahanovska YO, Sliusar MY, Bezrodnyi BH and Minchenko
OH: Hypoxic regulation of EDN1, EDNRA, EDNRB, and ECE1 gene
expressions in ERN1 knockdown U87 glioma cells. Endocr Regul.
53:250–262. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kawai K, Viars C, Arden K, Tarin D,
Urquidi V and Goodison S: Comprehensive karyotyping of the HT-29
colon adenocarcinoma cell line. Genes Chromosomes Cancer. 34:1–8.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Schust J, Sperl B, Hollis A, Mayer TU and
Berg T: Stattic: A small-molecule inhibitor of STAT3 activation and
dimerization. Chem Biol. 13:1235–1242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gatfield J, Mueller Grandjean C, Sasse T,
Clozel M and Nayler O: Slow receptor dissociation kinetics
differentiate macitentan from other endothelin receptor antagonists
in pulmonary arterial smooth muscle cells. PLoS One. 7:e476622012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sidharta PN, van Giersbergen PL, Halabi A
and Dingemanse J: Macitentan: Entry-into-humans study with a new
endothelin receptor antagonist. Eur J Clin Pharmacol. 67:977–984.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hong IS, Coe HV and Catanzaro LM:
Macitentan for the treatment of pulmonary arterial hypertension.
Ann Pharmacother. 48:538–547. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang J, Gareri C and Rockman HA:
G-protein-coupled receptors in heart disease. Circ Res.
123:716–735. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hauser AS, Attwood MM, Rask-Andersen M,
Schiöth HB and Gloriam DE: Trends in GPCR drug discovery: New
agents, targets and indications. Nat Rev Drug Discov. 16:829–842.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rosanò L, Cianfrocca R, Tocci P, Spinella
F, Castro VD, Caprara V, Semprucci E, Ferrandina G, Natali PG and
Bagnato A: Endothelin A receptor/β-arrestin signaling to the Wnt
pathway renders ovarian cancer cells resistant to chemotherapy.
Cancer Res. 74:7453–7464. 2014. View Article : Google Scholar
|
|
39
|
Kelland NF, Bagnall AJ, Morecroft I,
Gulliver-Sloan FH, Dempsie Y, Nilsen M, Yanagisawa M, Maclean MR,
Kotelevtsev YV and Webb DJ: Endothelial ET(B) limits vascular
remodelling and development of pulmonary hypertension during
hypoxia. J Vasc Res. 47:16–22. 2010. View Article : Google Scholar
|
|
40
|
Schneider MP, Boesen EI and Pollock DM:
Contrasting actions of endothelin ET(A) and ET(B) receptors in
cardiovascular disease. Annu Rev Pharmacol Toxicol. 47:731–759.
2007. View Article : Google Scholar
|
|
41
|
Wulfing C, Eltze E, Yamini J, Wülfing P,
Bierer S, Böcker W, Hertle L, Semjonow A and Sievert KD: Expression
of the endothelin axis in bladder cancer: Relationship to
clinicopathologic parameters and long-term survival. Eur Urol.
47:593–600. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fu W, Wu X, Yang Z and Mi H: The effect of
miR-124-3p on cell proliferation and apoptosis in bladder cancer by
targeting EDNRB. Arch Med Sci. 15:1154–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mahdi MR, Georges RB, Ali DM, Bedeer RF,
Eltahry HM, Gabr AEHZ and Berger MR: Modulation of the endothelin
system in colorectal cancer liver metastasis: Influence of
epigenetic mechanisms? Front Pharmacol. 11:1802020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hsu LS, Lee HC, Chau GY, Yin PH, Chi CW
and Lui WY: Aberrant methylation of EDNRB and p16 genes in
hepatocellular carcinoma (HCC) in Taiwan. Oncol Rep. 15:507–511.
2006.PubMed/NCBI
|
|
45
|
Tao K, Wu C, Wu K, Li W, Han G, Shuai X
and Wang G: Quantitative analysis of promoter methylation of the
EDNRB gene in gastric cancer. Med Oncol. 29:107–112. 2012.
View Article : Google Scholar
|
|
46
|
Yegnasubramanian S, Kowalski J, Gonzalgo
ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, Marzo AM, Isaacs WB
and Nelson WG: Hypermethylation of CpG islands in primary and
metastatic human prostate cancer. Cancer Res. 64:1975–1986. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Soh BS, Ng SY, Wu H, Buac K, Park JHC,
Lian X, Xu J, Foo KS, Felldin U, He X, et al: Endothelin-1 supports
clonal derivation and expansion of cardiovascular progenitors
derived from human embryonic stem cells. Nat Commun. 7:107742016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Alvarez D, Briassouli P, Clancy RM,
Zavadil J, Reed JH, Abellar RG, Halushka M, Fox-Talbot K, Barrat FJ
and Buyon JP: A novel role of endothelin-1 in linking Toll-like
receptor 7-mediated inflammation to fibrosis in congenital heart
block. J Biol Chem. 286:30444–30454. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cianfrocca R, Rosano L, Tocci P, Sestito
R, Caprara V, Castro VD, Maria RD and Bagnato A: Blocking
endothelin-1-receptor/beta-catenin circuit sensitizes to
chemotherapy in colorectal cancer. Cell Death Differ. 24:1811–1820.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wei W, Shi L, Chen W, Hu L, Chen D, Shi X,
Xiang H, Guo C and Wu Z: miR-200c regulates the proliferation,
apoptosis and invasion of gastric carcinoma cells through the
downregulation of EDNRA expression. Int J Mol Med. 41:1619–1626.
2018.
|
|
51
|
Sestito R, Cianfrocca R, Tocci P, Rosanò
L, Sacconi A, Blandino G and Bagnato A: Targeting endothelin 1
receptor-miR-200b/c-ZEB1 circuitry blunts metastatic progression in
ovarian cancer. Commun Biol. 3:6772020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Corvinus FM, Orth C, Moriggl R, Tsareva
SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal
K, et al: Persistent STAT3 activation in colon cancer is associated
with enhanced cell proliferation and tumor growth. Neoplasia.
7:545–555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C
and Lin J: STAT3 is necessary for proliferation and survival in
colon cancer-initiating cells. Cancer Res. 71:7226–7237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wendt MK, Balanis N, Carlin CR and
Schiemann WP: STAT3 and epithelial-mesenchymal transitions in
carcinomas. JAKSTAT. 3:e289752014.PubMed/NCBI
|
|
56
|
Thaete LG, Jilling T, Synowiec S, Khan S
and Neerhof MG: Expression of endothelin 1 and its receptors in the
hypoxic pregnant rat. Biol Reprod. 77:526–532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Morikawa T, Baba Y, Yamauchi M, Kuchiba A,
Nosho K, Shima K, Tanaka N, Huttenhower C, Frank DA, Fuchs CS and
Ogino S: STAT3 expression, molecular features, inflammation
patterns, and prognosis in a database of 724 colorectal cancers.
Clin Cancer Res. 17:1452–1462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kankanamalage SG, Karra AS and Cobb MH:
WNK pathways in cancer signaling networks. Cell Commun Signal.
16:722018. View Article : Google Scholar
|
|
59
|
Sato A, Shimizu M, Goto T, Masuno H,
Kagechika H, Tanaka N and Shibuya H: WNK regulates Wnt signalling
and β-Catenin levels by interfering with the interaction between
β-catenin and GID. Commun Biol. 3:6662020. View Article : Google Scholar
|
|
60
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhao J and Guan JL: Signal transduction by
focal adhesion kinase in cancer. Cancer Metastasis Rev. 28:35–49.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang Y, Wang Y, Che X, Hou K, Wu J, Zheng
C, Cheng Y, Liu Y, Hu X and Zhang J: Integrin alpha5 promotes
migration and invasion through the FAK/STAT3/AKT signaling pathway
in icotinib-resistant non-small cell lung cancer cells. Oncol Lett.
22:5562021. View Article : Google Scholar
|
|
63
|
Pei G, Lan Y, Chen D, Ji L and Hua ZC: FAK
regulates E-cadherin expression via p-SrcY416/p-ERK1/2/p-Stat3Y705
and PPARγ/miR-125b/Stat3 signaling pathway in B16F10 melanoma
cells. Oncotarget. 8:13898–13908. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pulido T, Adzerikho I, Channick RN,
Delcroix M, Galiè N, Ghofrani HA, Jansa P, Jing ZC, Brun FOL, Mehta
S, et al: Macitentan and morbidity and mortality in pulmonary
arterial hypertension. N Engl J Med. 369:809–818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Russignan A, Dal Collo G, Bagnato A,
Tamassia N, Bugatti M, Belleri M, Lorenzi L, Borsi E, Bazzoni R,
Gottardi M, et al: Targeting the endothelin-1 receptors curtails
tumor growth and angiogenesis in multiple myeloma. Front Oncol.
10:6000252020. View Article : Google Scholar
|
|
66
|
Tocci P, Cianfrocca R, Di Castro V, Rosanò
L, Sacconi A, Donzelli S, Bonfiglio S, Bucci G, Vizza E, Ferrandina
G, et al: β-arrestin1/YAP/mutant p53 complexes orchestrate the
endothelin A receptor signaling in high-grade serous ovarian
cancer. Nat Commun. 10:31962019. View Article : Google Scholar
|
|
67
|
Lee HJ, Hanibuchi M, Kim SJ, Yu H, Kim MS,
He J, Langley RR, Lehembre F, Regenass U and Fidler IJ: Treatment
of experimental human breast cancer and lung cancer brain
metastases in mice by macitentan, a dual antagonist of endothelin
receptors, combined with paclitaxel. Neuro Oncol. 18:486–496. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kim SJ, Lee HJ, Kim MS, Choi HJ, He J, Wu
Q, Aldape K, Weinberg JS, Yung WKA, Conrad CA, et al: Macitentan, a
dual endothelin receptor antagonist, in combination with
temozolomide leads to glioblastoma regression and long-term
survival in mice. Clin Cancer Res. 21:4630–4641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Damia G, Filiberti L, Vikhanskaya F,
Carrassa L, Taya Y, D'incalci M and Broggini M: Cisplatinum and
taxol induce different patterns of p53 phosphorylation. Neoplasia.
3:10–16. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Persons DL, Yazlovitskaya EM and Pelling
JC: Effect of extracellular signal-regulated kinase on p53
accumulation in response to cisplatin. J Biol Chem.
275:35778–35785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tchounwou PB, Dasari S, Noubissi FK, Ray P
and Kumar S: Advances in our understanding of the molecular
mechanisms of action of cisplatin in cancer therapy. J Exp
Pharmacol. 13:303–328. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ohtani H, Arimoto Y, Nishio K, Kanamiya Y,
Oba H, Adachi K, Shintani M, Nakamura R and Yui S: Efficacy and
toxicity of fluorouracil, leucovorin plus oxaliplatin (FOLFOX4 and
modified FOLFOX6) followed by fluorouracil, leucovorin plus
irinotecan(FOLFIRI)for advanced or metastatic colorectal
cancer-case studies. Gan To Kagaku Ryoho. 35:1769–1774.
2008.PubMed/NCBI
|
|
74
|
Gustavsson B, Carlsson G, Machover D,
Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F: A review of
the evolution of systemic chemotherapy in the management of
colorectal cancer. Clin Colorectal Cancer. 14:1–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|