Introduction
Numerous studies have reported an association
between psychological stress and the overall survival of patients
with various cancers, including breast, liver, lung, pancreatic,
prostate, colon and ovarian cancers (1–7).
Some studies have shown important cellular mechanisms that link the
stress responses of the hypothalamic-pituitary-adrenocortical (HPA)
axis or the sympathetic nervous system (SNS) with cancer
progression and metastasis (8–10).
Under conditions of psychological stress, the HPA axis is
activated, which leads to the release of corticotrophin-releasing
hormone from the hypothalamus and the secretion of
adrenocorticotrophic hormone from the anterior pituitary. As a
result, stress-associated hormones such as cortisol and
catecholamines including epinephrine (EPI) and norepinephrine (NE)
are released from the adrenal gland. These hormones exert strictly
controlled effects, including the elevation of blood pressure,
heart rate and blood sugar level, that prime an individual to
respond to a perceived threat. In addition to the HPA axis, the
central SNS is directly associated with the regulation of the
stress response via the release of catecholamines from autonomic
nerve endings (11). In contrast
to the transient and strictly controlled stress response, exposure
to sustained stress adversely affects tumor progression and cancer
therapy; dysregulation of the HPA axis has been shown to have a
detrimental effect on cancer incidence and survival. For example,
in one study, patients with hepatocellular carcinoma (HCC) had
significantly higher serum cortisol levels than healthy subjects
(12). In another study, patients
with breast cancer were reported to have high serum cortisol
levels, which could be suppressed by emotional support (13). Glucocorticoids can reach the tumor
microenvironment via the blood circulation and act on the
glucocorticoid receptors expressed by several types of cancer cells
to regulate diverse cellular signaling pathways. They thereby
increase the ability of cancer cells to proliferate and invade and
affect the interaction between cancer cells and their
microenvironment to promote tumor progression and metastasis
(14–16). Similarly, catecholamines can
contribute to stress-induced cancer growth and metastasis via
adrenergic receptors located on the surface of cancer cells,
leading to the activation of Ras/extracellular signal-regulated
kinase, NF-κB and cAMP-dependent protein kinase pathways, which
regulate cellular responses including proliferation,
differentiation and apoptosis (17,18).
In a study of women with triple-negative breast cancer treated with
neoadjuvant chemotherapy, the patients treated with β-blockers that
interfere with the receptor binding of EPI and other stress
hormones exhibited improved relapse-free survival compared with
women who did not receive β-blocker treatment (19). It is evident that surges in stress
hormone levels facilitate the development and progression of
various cancers.
Although there is considerable evidence associating
psychosocial stress with cancer development and progression, the
mechanism is not fully understood. One possible mechanism is that
exposure to stress and stress hormones such as cortisol and
catecholamines increases DNA damage and promotes tumorigenesis
(20,21). A study demonstrated that the
incubation of 3T3 mouse fibroblasts with EPI or NE resulted in
long-term DNA damage that was sufficient to induce genomic
instability and vulnerability to tumor transformation (22). Moreover, another study revealed
that physiological concentrations of cortisol induced DNA damage in
cells, which was associated with the transcriptional upregulation
of DNA damage sensors, including checkpoint kinase 1 (Chk1) and
Chk2 and the cell cycle regulator gene cell division cycle 25A
(20). Consequently, long-term
effects on genomic stability result in increased cell
transformation and/or tumor formation. Additionally, numerous
studies have demonstrated that stress increases the growth of
existing tumors and the risk of tumor metastasis (23–25).
Proposed mechanisms for these stress effects include increases in
VEGF and angiogenesis resulting from the activation of β-adrenergic
pathways (26). Stress hormones
have also been shown to promote the migration and invasion of
malignant cells via the elevation of matrix metalloproteinases,
which are known for their ability to degrade the extracellular
matrix and facilitate cell invasion (27,28).
Hence, stress contributes to the initiation, growth and metastasis
of tumors.
However, it appears contradictory that the stress
response induces DNA damage but also promotes the growth and
metastasis of existing tumors. We hypothesize that cancer cells
undergo DNA damage adaptation to survive stimulation by chronic
stress; DNA damage adaptation is a process by which cancer cells
adapt to DNA damage and allow cell division despite the presence of
unrepaired DNA damage (29,30).
Various oncogenic signaling pathways are also activated to promote
uncontrolled tumor growth in this process. The adaptation to DNA
damage is mediated by checkpoint recovery process effectors, which
are considered oncogenes. Wild-type p53-induced phosphatase 1
(Wip1), also known as protein phosphatase,
Mg2+/Mn2+ dependent 1D or protein phosphatase
2Cδ, is a type 2C family serine/threonine phosphatase that
contributes to the inactivation of checkpoint recovery by removing
DNA damage-induced phosphorylation in several of its components,
including p53, ataxia-telangiectasia mutated (ATM), ataxia
telangiectasia and Rad3 related (ATR), Chk1, Chk2 and
phosphorylated histone 2AX (γ-H2AX) (31). Wip1 was first identified as a p53
target gene, which negatively regulates the function and stability
of p53 following cellular stress (32). In a study of Fanconi anemia, a
chromosomal instability syndrome characterized by bone marrow
failure and a predisposition to cancer, Fanconi anemia cells with a
large amount of DNA damage continued to undergo cell division after
the induction of DNA damage by ignoring the presence of unrepaired
DNA damage, while the inhibition of Wip1 prevented cell cycle
progression and division (33).
Therefore, we hypothesize that the upregulation of Wip1 preserves
the capacity of cells to divide when stress hormones induce DNA
damage in cancer cells.
Liver cancer is among the most common cancers in
China and >50% of the liver cancer cases and deaths worldwide
are estimated to occur in China (34). However, the effects of stress
response on liver cancer are yet to be determined. A previous study
demonstrated that the upregulation of Wip1 expression is associated
with progressive pathological features and a poor prognosis in
patients with HCC (35). In
addition, Xu et al reported that Wip1 was highly expressed
in ~59% of patients with HCC and its upregulation was an
independent predictor of HCC-specific overall survival (36). With the aim of investigating the
impact of stress hormones on the survival of liver cancer cells,
the present study examined the expression of Wip1 in HepG2 cells
treated by stress hormones and identified the role of Wip1 in the
DNA damage and apoptosis induced by stress hormone stimulation.
Materials and methods
Chemicals and reagents
Dexamethasone (DEX), EPI and NE were purchased from
Target Molecule Corp. All compounds were dissolved in dimethyl
sulfoxide (DMSO) to a concentration of 10 or 20 mM and preserved at
−20°C. DMSO served as the vehicle control in all experiments. The
antibody against Wip1 was obtained from Abcam (cat. no. ab31270;
dilution 1:1,000). Antibodies against ATM (cat. no. 2873),
phosphorylated (p-)ATM (Ser1981) (cat. no. 5883), Chk2 (cat. no.
2662), p-Chk2 (Thr68) (cat. no. 2197), p-p53 (Ser15) (cat. no.
9287), p53 (cat. no. 9282), γ-H2AX (cat. no. 9718) and H2AX (cat.
no. 7631), and a second primary antibody against Wip1 (cat. no.
11901) were purchased from Cell Signaling Technology, Inc.
Antibodies against β-actin (cat. no. AC026) and GAPDH (cat. no.
AC033) were obtained from ABclonal Biotech Co., Ltd. All antibodies
were used at the dilutions recommended by the manufacturer unless
otherwise specified.
Cell culture and cell viability
assay
The HepG2 human liver cancer cell line (a gift from
Dr Jingrong Cui at Peking University, Beijing, China) was cultured
in Eagle's minimum essential medium (Macgene™; M&C Gene
Technology) supplemented with 10% (v/v) fetal bovine serum (FBS;
PAN-Biotech GmbH), streptomycin (100 µg/ml; Macgene; M&C Gene
Technology) and penicillin (100 U/ml; Macgene; M&C Gene
Technology) in a 5% CO2 incubator at 37°C. To determine
the cytotoxicity of DEX, EPI and NE, HepG2 cells were treated with
or without each of these compounds for 72 h in a 5% CO2
incubator at 37°C followed by a cell viability assay using Cell
Counting Kit-8 (Dojindo Laboratories, Inc.) according to the
manufacturer's instructions.
Transient transfection
The small interfering RNAs (siRNAs), namely Wip1
siRNA (cat. no. sc-39205) and control siRNA (cat. no. sc-36869 or
sc-37007), were purchased from Santa Cruz Biotechnology, Inc.
Briefly, 40 µl of 10 µM siRNA duplex was diluted with 400 µl siRNA
Transfection Medium (Santa Cruz Biotechnology, Inc.) and then mixed
with 30 µl siRNA Transfection Reagent in 400 µl siRNA Transfection
Medium followed by incubation for 40 min at room temperature. In a
10-cm tissue culture dish of ~80% confluent cells, each
transfection was conducted by washing with 2 ml of siRNA
Transfection Medium and adding 3.2 ml siRNA Transfection Medium and
800 µl transfection mixture. After incubation for 5 h at 37°C in a
5% CO2 incubator, cells were supplemented with 4 ml
normal growth medium containing two times the normal serum and
antibiotics concentration without removing the transfection
mixture, and incubated for an additional 18 h followed by cell
reseeding for the subsequent experiments.
Alkaline comet assay
The alkaline comet assay was performed using the
Trevigen CometAssay® Kit (Trevigen, Inc.; Bio-Techne)
according to the manufacturer's instructions. Briefly, HepG2 cells
were seeded at 8×104 cells/well in a 12-well plate.
After 24 h, the cells were treated with vehicle control, 5 µΜ DEX,
1 µΜ EPI or 1 µΜ NE for 4 h in a 5% CO2 incubator at
37°C. Then the cells were harvested, washed twice with
phosphate-buffered saline (PBS) and mixed with 1% low-melting
agarose in PBS at 37°C to form a single cell suspension embedded in
agarose. The resulting mixture was immediately pipetted on with the
sample area of a CometSlide (Trevigen, Inc.; Bio-Techne). The
slides were placed at 4°C for 10 min and then lysed at 4°C
overnight in the dark. After lysis, the slides were subjected to
electrophoresis at 21 V for 45 min at 4°C, and then immersed twice
in distilled water for 5 min and once in 70% (v/v) ethanol for 5
min. The slides were dried completely at 37°C for 10–15 min and
then stained with propidium iodide (PI) for 10 min at room
temperature. Comets were observed using an Olympus BX53
fluorescence microscope (Olympus Corp.) equipped with an Olympus
DP72 camera (Olympus Corp.). The percentage of DNA in the tail was
quantified based on ≥30 randomly selected cells in each sample
using Image J software (version 1.53c; National Institutes of
Health) as previously described (37).
Cell apoptosis assay
HepG2 cells were seeded at 2×105
cells/well into a 6-well plate. After 24 h, the cells were treated
with vehicle control, 5 µΜ DEX, 1 µΜ EPI or 1 µΜ NE for 48 h in a
5% CO2 incubator at 37°C. To quantify apoptosis, the
treated cells were collected and stained with Annexin V-FITC and PI
using an Annexin V/PI Apoptosis Detection Kit (Dojindo
Laboratories, Inc.) according to the supplier's instructions as
previously described (38,39). The apoptotic cells were then
analyzed using a BD Accuri™ C6 Flow Cytometer (BD Biosciences) and
built-in data analysis software (version 1.0.264.15; BD
Biosciences).
Immunofluorescence staining
HepG2 cells were cultured on coverslips in a 6-well
plate. After exposure to vehicle control, DEX, EPI or NE for 4 or
48 h, the cells were washed with PBS and fixed with 4% formaldehyde
in PBS at room temperature for 15 min. The fixed cells were washed
with 0.2% Triton X-100 at 4°C for 2 min, incubated with ice-cold
methanol at −20°C for 10 min. After rinsing with PBS at room
temperature for 5 min, the cells were blocked with 10% blocking
serum (Beijing Solarbio Science & Technology Co., Ltd.) in PBS
at 37°C for 30 min. The cells were then probed with anti-γ-H2AX
antibody (1:200 dilution) overnight at 4°C followed by washing with
PBS containing 1% BSA and incubation with FITC-conjugated goat
anti-rabbit IgG (1:50 dilution; cat. no. ZF-0311; ZSGB Biotech) at
room temperature for 1 h. After washing thrice with 1% BSA (cat.
no. AP0027; NOVON Scientific) in PBS, the coverslips were incubated
with 4′,6-diamidine-2-phenylindole dihydrochloride (300 nM in PBS)
for 30 min in the dark. The coverslips were then washed thrice with
distilled water and mounted on slides before viewing with an
Olympus DP72 microscope (Olympus Corp.).
Cycloheximide chase assay
A cycloheximide chase assay was performed to analyze
protein stability as previously described (40). A total of 1×106 HepG2
cells were plated in a 10-cm tissue culture dish. After 24 h, the
cells were cotreated with 10 µg/ml cycloheximide (cat. no. C7698;
MilliporeSigma) in the presence of DMSO vehicle control, 5 µΜ DEX,
1 µΜ EPI or 1 µΜ NE for 0, 6, 12, 24, 36, 48, 60 and 72 h in a 5%
CO2 incubator at 37°C, followed by cell lysate
preparation and western blotting analysis of Wip1 expression.
Western blotting
Western blotting analysis was performed to determine
protein expression levels as previously described (41,42).
Following exposure to vehicle control, DEX, EPI or NE, HepG2 cells
were harvested and lysed using TNN buffer (50 mM Tris·HCl, pH 7.4;
150 mM NaCl; 20 mM EDTA; 0.5% NP-40; 1 mM
Na3VO4; 50 mM NaF) with 2 mM PMSF and 1 mM
dithiothreitol. After incubation on ice for 10 min and brief
sonication, the cell lysates were centrifuged at 14,000 × g at 4°C
for 30 min and the supernatants were collected. The concentration
of protein in the supernatant was quantified using a Bicinchoninic
Acid Protein Assay Kit (Beijing Dingguo Changsheng Biotechnology
Co., Ltd.). Equal amounts (40–80 µg) of the protein lysates were
separated using 8–15% sodium dodecyl sulfate polyacrylamide gel
electrophoresis. Then, the proteins were electroblotted onto
polyvinylidene fluoride membranes (MilliporeSigma). The membranes
were blocked in 5% non-fat milk in Tris-buffered saline containing
0.1% Tween-20 (TBST) for 60 min followed by incubation with primary
antibodies against different proteins at 4°C overnight. After
washing with TBST three times, the membranes were probed with
secondary anti-IgG antibody conjugated with horseradish peroxidase
[cat. no. 7074 (anti-rabbit) and 7076 (anti-mouse); 1:2,000
dilution; Cell Signaling Technology, Inc.] for 1 h at room
temperature and visualized using an Efficient Chemiluminescence Kit
(cat. no. GE2301-100ML; Beijing Dingguo Changsheng Biotechnology
Co., Ltd.) with a Tanon Imaging System (Tanon Science &
Technology Co., Ltd.). The relative protein levels were determined
by measuring the intensity of the bands using Gel-Pro Analyzer
software (version 4.0.00.001; Media Cybernetics, Inc.) and the
target proteins were normalized against the internal control, GAPDH
or β-actin.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cultured cells
using E.Z.N.A.® Total RNA Kit I (Omega Bio-Tek, Inc.) as
described previously (43). The
concentration of total RNA was measured using a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). The synthesis of cDNA was performed using an All-In-One 5X
RT MasterMix kit (Applied Biological Materials, Inc.) according to
the manufacturer's recommendations. qPCR was then performed using
EvaGreen® qPCR MasterMix (Applied Biological Materials,
Inc.) on a StepOnePlus™ Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The primers were used as follows:
Wip1 forward, 5′-CTGTACTCGCTGGGAGTGAG-3′ and reverse,
5′-GTTCGGGCTCCACAACGATT-3′; and GAPDH forward,
5′-AAGGACTCATGACCACAGTCCAT-3′ and reverse,
5′-CCATCACGCCACAGTTTCC-3′. GAPDH was used as the internal control.
Thermocycling was initiated by a 10-min incubation at 95°C,
followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The
relative mRNA levels of Wip1 were determined using the
2−ΔΔCq method (44).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. To assess the differences among groups, statistical
analyses were performed by one-way analysis of variance (ANOVA)
followed by Dunnett's post hoc test. For analyses with two
independent variables, the data were analyzed by two-way ANOVA
followed by Tukey's post hoc test. The analyses were performed
using SPSS software (version 21; IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Stress hormones induce DNA damage in
HepG2 human liver cancer cells
Exposure to stress or stress hormones has been
reported to alter the genetic integrity of cancers by damaging
their DNA and/or influencing the DNA maintenance machinery,
specifically DNA damage response and DNA repair (20,21).
To assess the DNA damage of HepG2 cells following exposure to
stress hormones including cortisol and catecholamines, an alkaline
comet assay was performed. The concentrations of DEX, EPI and NE
were selected based on previous studies of stress hormone-mediated
responses in HepG2 cells (45–49).
A cytotoxicity assay also confirmed that the compounds were not
cytotoxic at the concentrations used (Fig. S1). The alkaline comet assay was
performed to quantify DNA damage after the exposure of HepG2 cells
to 5 µM DEX, 1 µM EPI or 1 µM NE. A time-course experiment firstly
verified that distinct DNA damage occurred 4 h following treatment
with 1 µM NE (Fig. S2). When
HepG2 cells were treated with 5 µM DEX for 4 h, the percentage of
DNA in the comet's tail exhibited a statistically significant
increase in DNA damage in response to DEX compared with
vehicle-treated cells (32 vs. 6%; Fig.
1A and B). Treatment with 1 µM EPI or NE also induced similarly
elongated tails compared with the vehicle control (38 and 28%,
respectively; Fig. 1A and B). The
DNA damage response persisted when the treatment duration was
prolonged to 48 h (Fig. 1A and B).
The DEX-treated HepG2 cells exhibited 2.7-fold more DNA damage
compared with the DMSO vehicle-treated cells. Persistent DNA
fragmentation was also observed in the EPI and NE-treated cells
(1.6- and 3.0-fold higher than that in the vehicle-treated cells,
respectively, although the increase was not significant for EPI).
These results suggest that stress hormones induce DNA damage in
HepG2 cells. To verify these findings, the level of γ-H2AX, an
indicator of DNA damage, was evaluated by immunofluorescence in
HepG2 cells following treatment with 5 µM DEX, 1 µM EPI or 1 µM NE
for 4 and 48 h. The results showed increases in the punctate
staining of γ-H2AX in the nuclei of HepG2 cells at 4 h after the
stress hormone treatments compared with that in the vehicle-treated
control cells (Fig. 1C). At 48 h
after the treatments, the nuclear staining of γ-H2AX in the treated
HepG2 cells remained at high levels (Fig. 1C). These observations are
consistent with the findings of the comet assay.
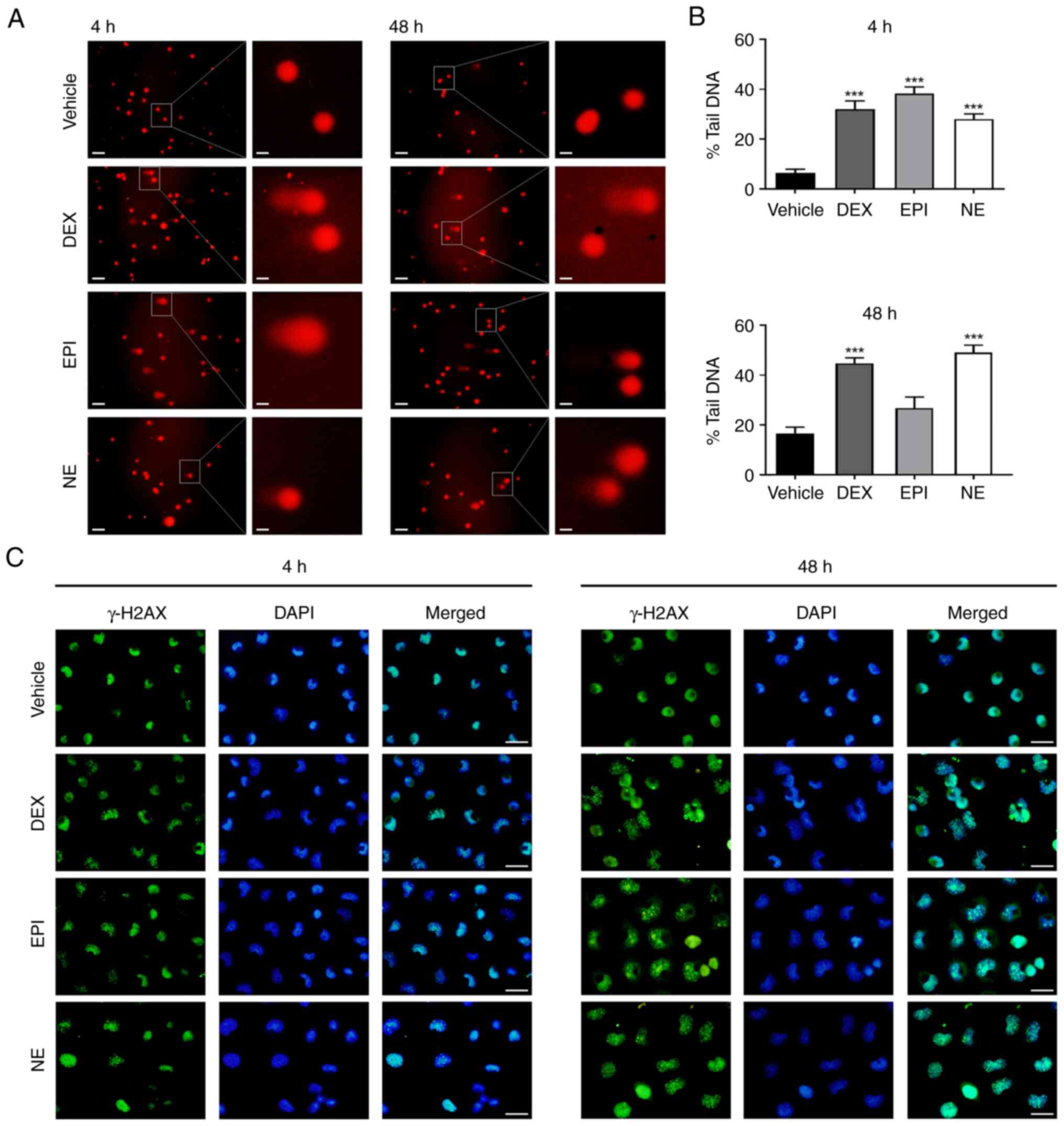 | Figure 1.Stress hormones induce DNA damage in
HepG2 cells. (A) Representative images of the comet assay. HepG2
cells were treated with DMSO vehicle control, 5 µM DEX, 1 µM EPI or
1 µM NE for 4 or 48 h and then subjected to an alkaline comet
assay. Scale bar, 25 µm for main images and 5 µm for insets. (B)
Histograms showing a quantitative analysis of the percentage of DNA
in the comet tail after treatment with DEX, EPI or NE for 4 or 48
h. (C) Immunofluorescence staining of γ-H2AX in HepG2 cells
following treatment with DMSO vehicle control, 5 µM DEX, 1 µM EPI
or 1 µM NE for 4 or 48 h. Cell nuclei were stained with DAPI. Scale
bar, 25 µm. ***P<0.001 vs. vehicle control. DMSO,
dimethylsulfoxide; DEX, dexamethasone; EPI, epinephrine; NE,
norepinephrine; γ-H2AX, phosphorylated histone 2AX. |
HepG2 cells adapt to stress
hormone-induced DNA damage
DNA damage activates DNA damage response factors
which activate p53 to allow the accumulation of apoptotic proteins,
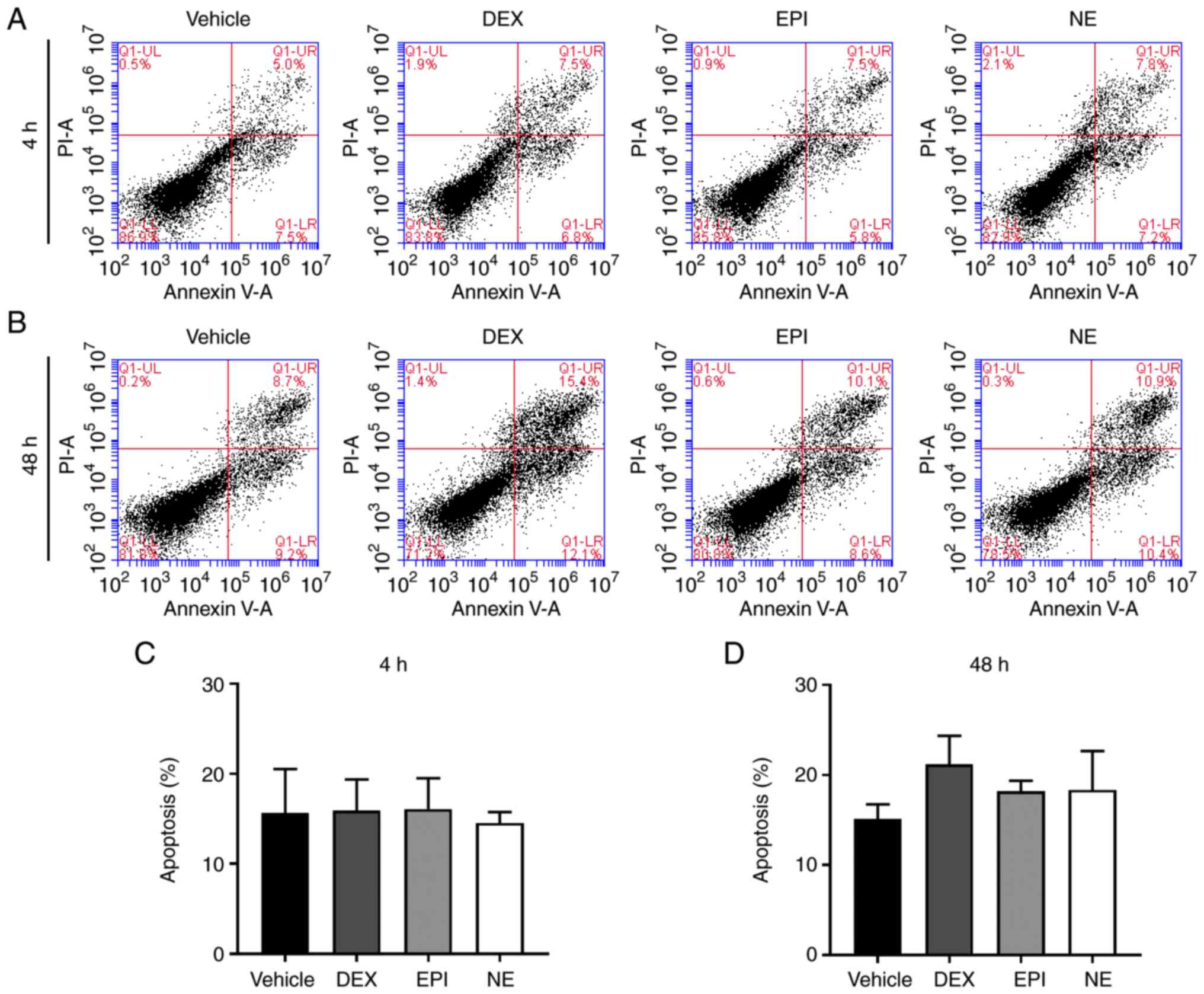
thereby triggering cell death. In order to investigate this, a flow
cytometric analysis was performed to quantify the proportion of
apoptotic cells following treatment with DEX, EPI or NE. As shown
in Fig. 2, no significant increase
in apoptosis occurred in HepG2 cells after treatment with DEX, EPI
and NE for 4 or 48 h when compared with the vehicle-treated
control. These findings suggest that HepG2 cells adapt to the
damage induced by stress hormones despite the presence of DNA
lesions.
Stress hormones dephosphorylate DNA
damage response factors by the upregulation of Wip1 in HepG2
cells
To identify how cancerous cells escape stress
hormone-induced DNA stress, the levels of DNA damage response
factors were determined in HepG2 cells following treatment with
DEX, EPI or NE using western blotting. Fig. 3 show that the three stress hormones
induced increments in the p-ATM (Ser1981)/ATM and p-Chk2
(Thr68)/Chk2 ratios compared with those in the cells treated with
vehicle control following a 4-h treatment, indicative of the
occurrence of stress hormone-induced DNA damage. These observations
are consistent with the results of the comet assay. It is well
established that Wip1 plays a vital role in stress signaling and
the DNA damage response. When external stress induces DNA damage,
Wip1 negatively regulates the DNA damage response via the
dephosphorylation of its target proteins, including p53, MAPK and
NF-κB. On the basis that stress hormones induce DNA damage instead
of killing cells, we hypothesized that Wip1 negatively modulates
the stress hormone-induced DNA damage response and allows tumor
cells to adapt to the DNA damage. Therefore, the expression of Wip1
was examined following the various stress hormone treatments. As
shown in Fig. 3, the expression of
Wip1 increased following treatment with DEX, EPI or NE. The Wip1
expression level was observed to rise 48 h after DEX treatment. EPI
induced a significant elevation at 4 h while NE significantly
increased Wip1 expression after 4 h and further increased it at 48
h. However, the elevation of p-ATM and p-Chk2 levels was abrogated
after 48 h. These findings suggest that Wip1 has a negative
regulatory effect on stress hormone-induced DNA damage.
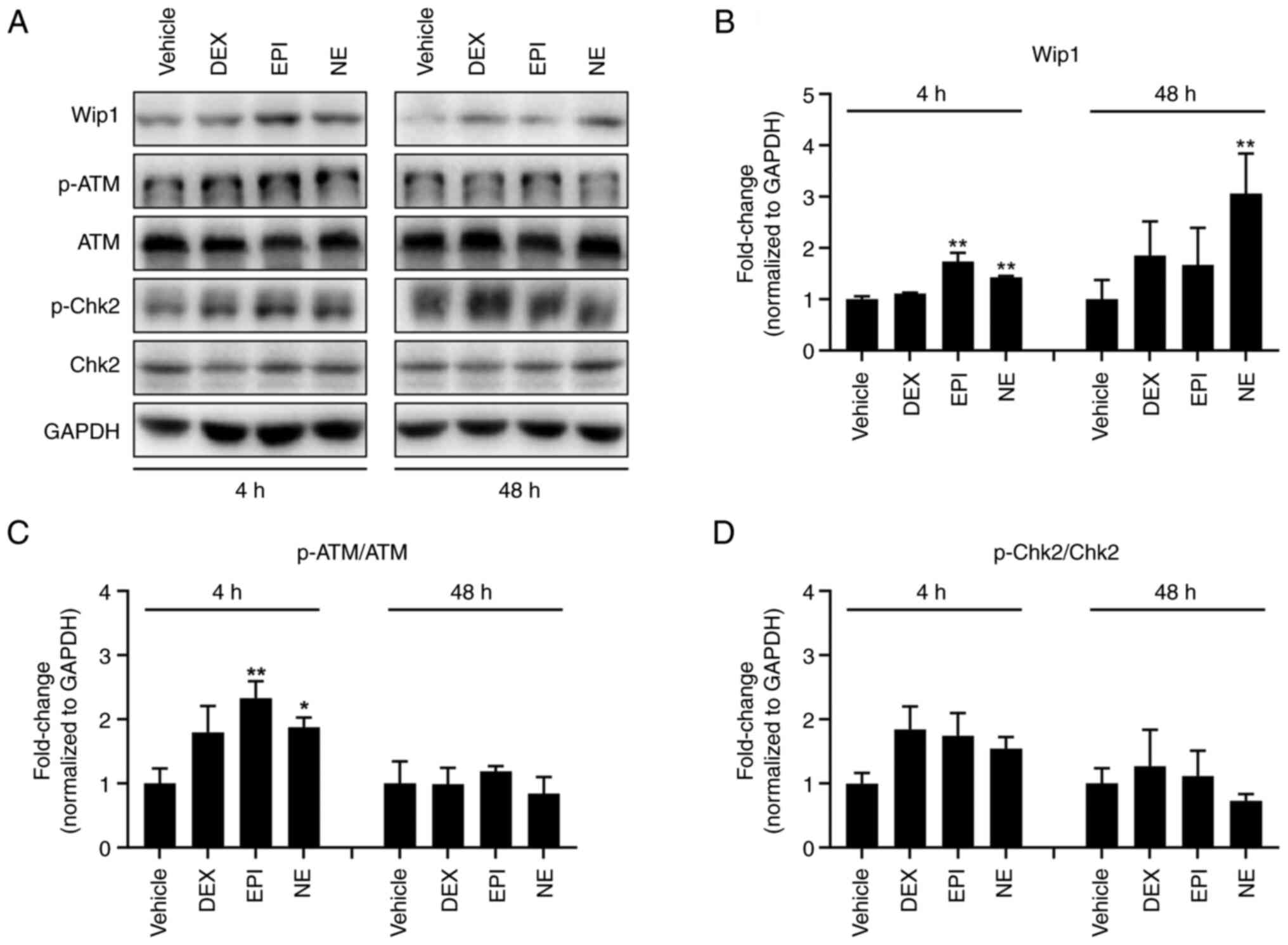 | Figure 3.Stress hormones upregulate the Wip1
protein level in HepG2 cells. (A) Western blotting analysis of
factors involved in the DNA damage response of HepG2 cells
following treatment with DMSO vehicle control, 5 µM DEX, 1 µM EPI
or 1 µM NE for 4 or 48 h. GAPDH was used as a loading control.
Specific protein bands were determined according to their molecular
weight. Note that the smearing and double banding of p-Chk2 may be
due to low antibody specificity and future experiments may benefit
from a change of antibody. Bar charts show quantitative data for
(B) Wip1, (C) p-ATM and (D) p-Chk2 based on measurements of the
density of the western blot bands and normalized against the GAPDH
internal control or the total protein. *P<0.05 and **P<0.01
vs. the DMSO vehicle control. Wip1, wild-type p53-induced
phosphatase 1; DMSO, dimethylsulfoxide; DEX, dexamethasone; EPI,
epinephrine; NE, norepinephrine; p-, phosphorylated; ATM,
ataxia-telangiectasia mutated; Chk2, checkpoint kinase 2. |
Stress hormones enhance Wip1 protein
stability in HepG2 cells
To investigate the mechanism by which stress
hormones upregulate Wip1, RT-qPCR was performed in HepG2 cells and
the Wip1 mRNA level was observed to be unchanged following stress
hormone treatment (Fig. 4A). Since
no significant change was detected in the Wip1 mRNA level following
the treatment of HepG2 cells with DEX, EPI or NE for 48 h, we
hypothesized that the mechanism of stress hormone-mediated Wip1
upregulation is a post-transcriptional process and examined the
possibility that stress hormones maintain the stability of Wip1. To
test this possibility, HepG2 cells were co-treated with
cycloheximide, which is an inhibitor of elongation during protein
synthesis, and DEX, EPI or NE; the effect on Wip1 protein stability
was then determined. As is evident from Fig. 4B-E, the vehicle-treated cells
exhibited a significant decline in Wip1 protein level from 24 h
following cycloheximide treatment. However, when the HepG2 cells
were co-treated with DEX and cycloheximide, Wip1 did not
significantly decrease until 60 h after the start of treatment. The
Wip1 protein level in the control cells was also lower than that of
EPI- or NE-treated cells upon cycloheximide treatment while a
significant loss of Wip1 in the cells co-treated with EPI or NE was
observed at 72 or 60 h, respectively, following cycloheximide
treatment. Overall, the loss of Wip1 in HepG2 cells treated with a
combination of stress hormone and cycloheximide was much slower
than that of the control treatment without stress hormones. This
indicates that stress hormones maintain the stability of Wip1
protein.
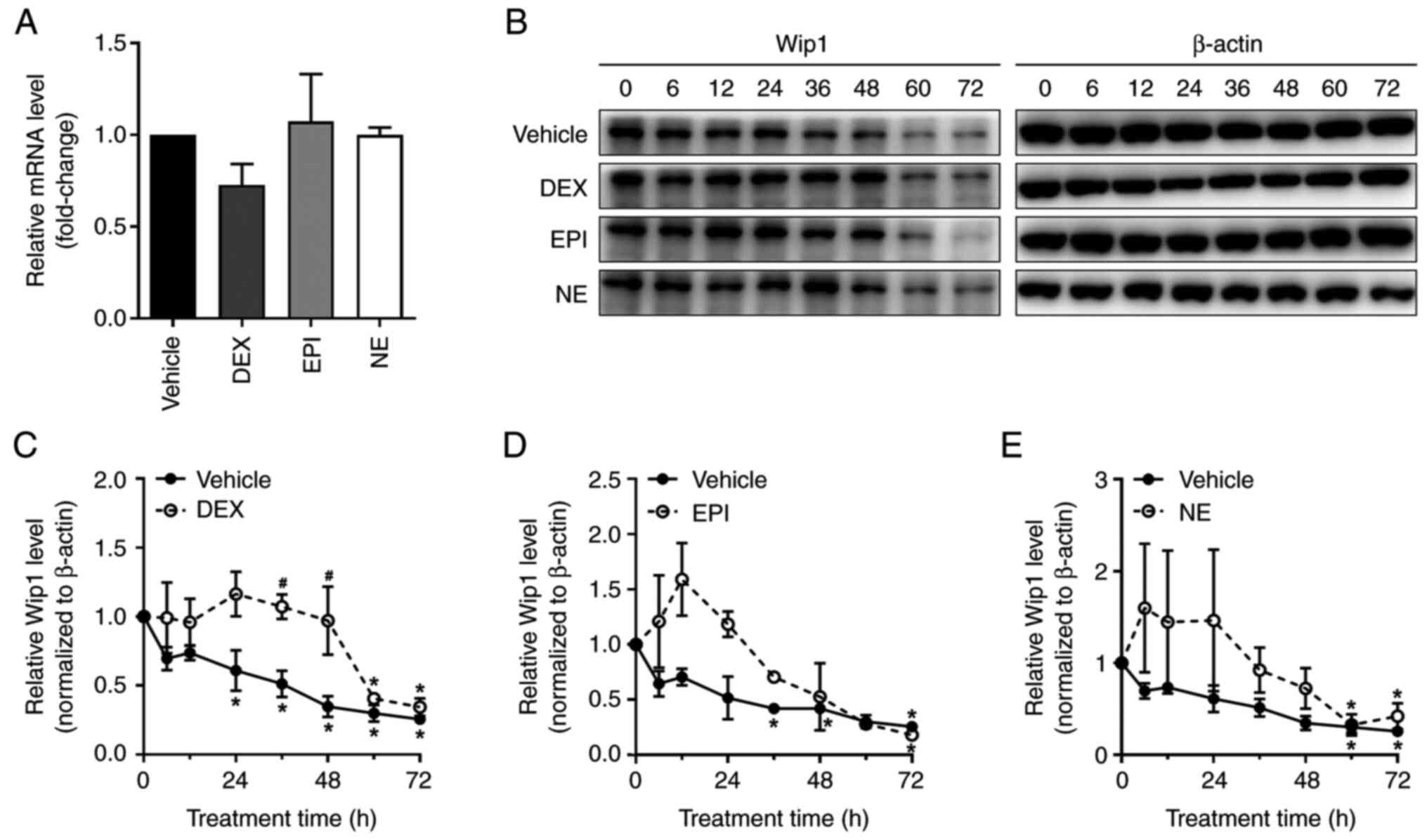 | Figure 4.Stress hormones enhance the stability
of Wip1 protein in HepG2 cells. (A) HepG2 cells were treated with
DMSO vehicle control, 5 µM DEX, 1 µM EPI or 1 µM NE for 48 h and
harvested for the analysis of Wip1 mRNA expression using reverse
transcription-quantitative polymerase chain reaction. (B) HepG2
cells were treated with DMSO vehicle control, 5 µM DEX, 1 µM EPI or
1 µM NE for various time points in the absence or presence of 10
µg/ml cycloheximide and subjected to the analysis of Wip1
expression by western blotting. The Wip1 levels of the western
blots for cells treated with (C) DEX, (D) EPI and (E) NE were
determined by measuring the intensity of the bands using
densitometric software, normalized against the internal control
β-actin and plotted against the time of treatment. Data shown are
the mean ± standard deviation of three independent experiments.
*P<0.05 vs. 0 h; #P<0.05 vs. vehicle control at
the corresponding time point. Wip1, wild-type p53-induced
phosphatase 1; DMSO, dimethylsulfoxide; DEX, dexamethasone; EPI,
epinephrine; NE, norepinephrine. |
Wip1 knockdown increases the levels of
DNA damage response factors following treatment with stress
hormones
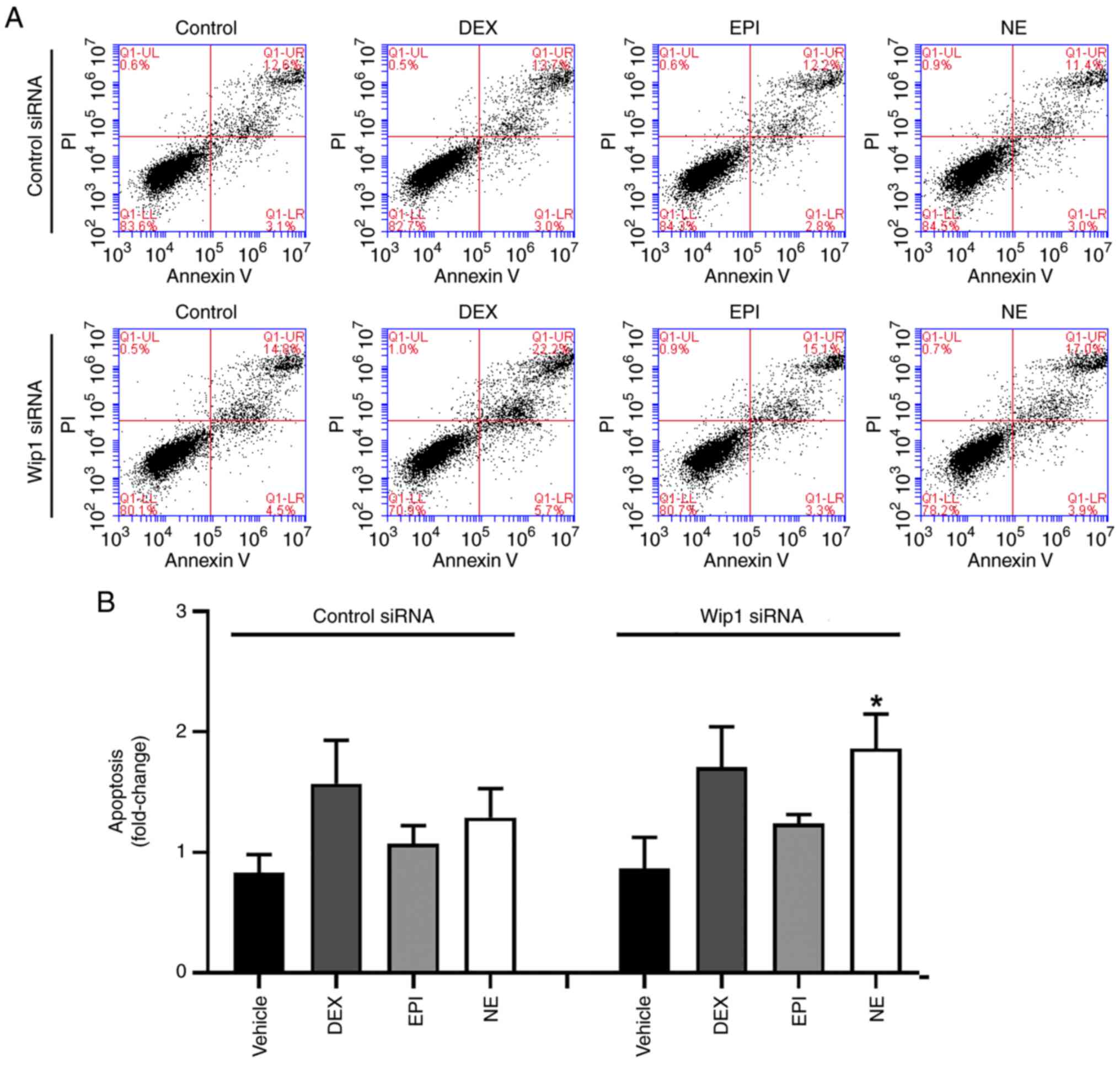
To explore the role of Wip1 in the regulation of
stress hormone-induced DNA damage, Wip1 protein expression in HepG2
cells was depleted using siRNA. Optimal transfection conditions
were firstly identified via the use of different ratios of Wip1
siRNA and transfection reagents (Fig.
S3). The siRNA-mediated knockdown of Wip1 in the HepG2 cells
was demonstrated by the reduced expression level of Wip1 compared
with that in cells transfected with control siRNA (Fig. 5A). Next, whether the loss of Wip1
affects stress hormone-induced DNA damage was examined via the
immunoblotting of DNA damage response factors. Following treatment
with stress hormones for 4 h, the Wip1-knockdown HepG2 cells
presented elevated levels of γ-H2AX, a sensor of DNA double-strand
breaks, indicating the occurrence of DNA damage (Figs. 5B and S4). When DNA damage occurs, the
phosphorylation of Chk2 at Thr68 by ATM is attributable to the
activation of Chk2 and downstream signaling (50–52).
Also, DNA damage induces the phosphorylation p53 at Ser15 and
Ser20, leading to cell cycle arrest, DNA repair or apoptosis
(53,54). In the present study, the levels of
p-p53 (Ser15) and p-Chk2 (Thr68) were also increased in
Wip1-knockdown HepG2 cells, although the difference observed was
not found to be significant due to high variation among the results
of different experiments (Figs. 5B
and S4). These findings suggest
that Wip1 is involved in the DNA damage induced by stress hormones.
To further verify the above findings, the level of γ-H2AX in HepG2
cells was determined by immunofluorescence 48 h after the knockdown
of Wip1. The knockdown of Wip1 attenuated the punctate staining of
γ-H2AX in the nuclei of HepG2 cells at 48 h after treatment
compared with that in the respective cells transfected with control
siRNA (Fig. 5C). Finally, the role
of Wip1 in stress hormone-induced apoptosis was determined in
Wip1-knockdown HepG2 cells following treatment with DEX, EPI or NE
for 48 h. As is evident from Fig.
6, an increase in the proportion of apoptotic cells was
observed in Wip1-knockdown HepG2 cells after stress hormone
treatment, suggesting that Wip1 serves a role in the protection of
HepG2 cells from stress hormone-induced apoptosis. However, this
evidence requires further verification.
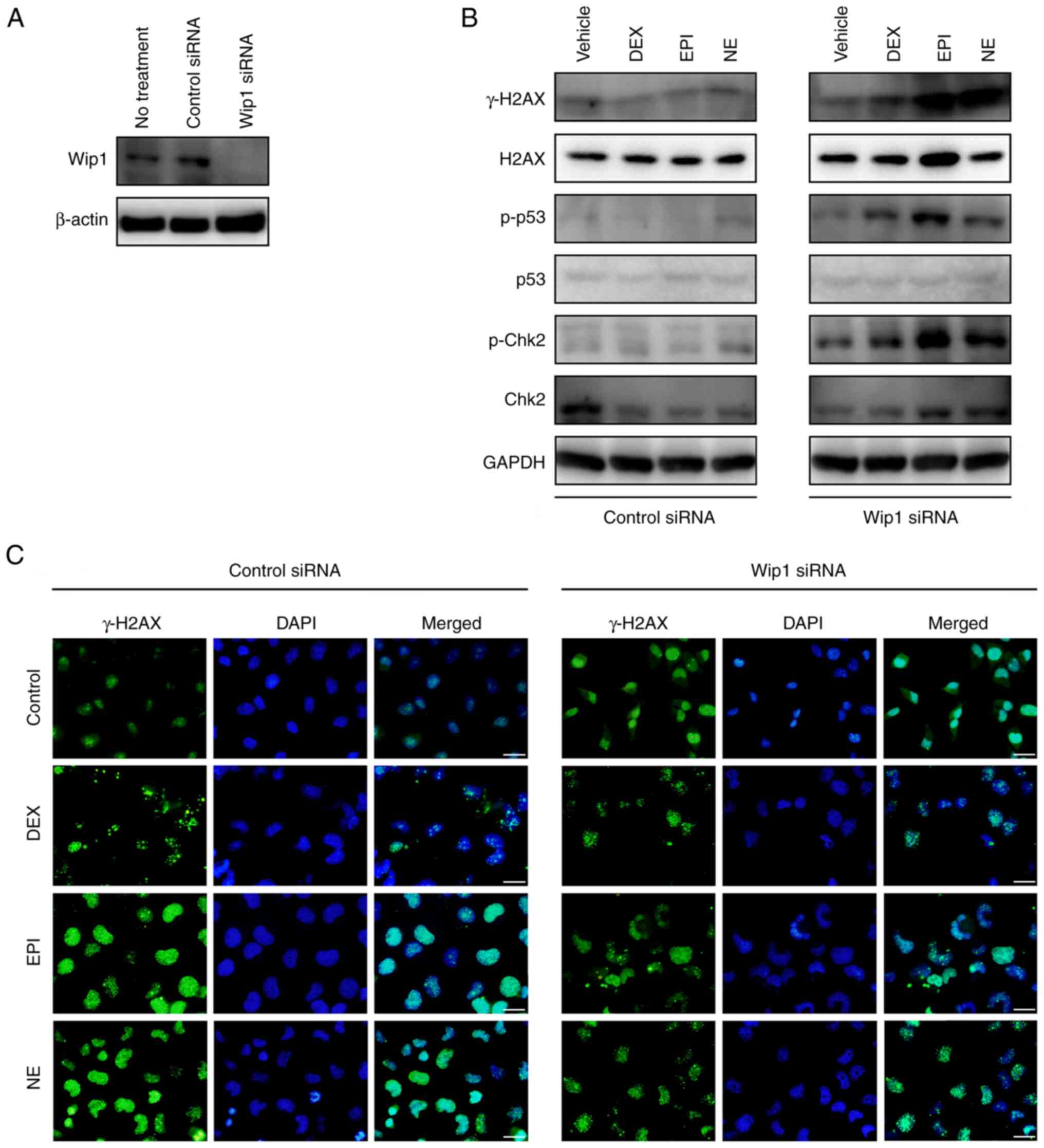 | Figure 5.Wip1 knockdown cells are more
sensitive to stress hormone-induced DNA damage response. (A)
Western blotting assay of the Wip1 protein level in HepG2 cells
transfected with control siRNA or Wip1 siRNA for 48 h. (B) Western
blot analysis of DNA damage response factors in Wip1 knockdown and
control cells following treatment with DMSO vehicle control, 5 µM
DEX, 1 µM EPI or 1 µM NE for 4 h. GAPDH was used as a loading
control. Specific protein bands were determined according to their
molecular weight. Note that the smearing and double banding of
p-Chk2 may be due to low antibody specificity and future
experiments may benefit from a change of antibody. (C)
Immunofluorescence staining of γ-H2AX in Wip1 knockdown and control
HepG2 cells following treatment with DMSO vehicle control, 5 µM
DEX, 1 µM EPI or 1 µM NE for 48 h. Cell nuclei were stained with
DAPI. Scale bar, 25 µm. Wip1, wild-type p53-induced phosphatase 1;
siRNA, small interfering RNA; DMSO, dimethylsulfoxide; DEX,
dexamethasone; EPI, epinephrine; NE, norepinephrine; H2AX, histone
2AX; γ-H2AX, phosphorylated H2AX; p-, phosphorylated; Chk2,
checkpoint kinase 2. |
Discussion
In the present study, a DNA damage response was
observed following the treatment of HepG2 cells with DEX, EPI and
NE, but the induction of apoptotic cell death did not occur. Wip1,
a type 2C family serine/threonine phosphatase, was found to
contribute to the adaption of stress hormone-induced DNA damage by
an upregulation in expression resulting from enhanced protein
stability. The results of the present explorative study suggest
that HepG2 cells may escape the stress hormone-induced DNA damage
response via the suppression of DNA damage response factors.
Ultimately, cancerous cells may take advantage of DNA damage
adaptation to survive stimulation by chronic stress; however, the
in-depth mechanism requires prospective validation.
Cancer research has predominantly focused on
investigations of gene mutations and signal transduction pathways
in tumor cells, whereas the potential role of phycological stress
has been less fully explored. The effects of stress hormones on
tumor growth are mediated via the wide distribution of stress
hormone receptors in cancer cells and the cellular signaling that
occurs in response to the binding of stress hormones to their
receptors. A variety of cancer cells such as liver, breast and
ovarian cancer cells express receptors for glucocorticoids from the
HPA axis and catecholamines from the SNS (55–57).
The overexpression of glucocorticoid receptors has been identified
as a potent survival pathway in breast cancer cells, and a study
revealed that glucocorticoid receptor-mediated cancer cell survival
signaling was activated by the administration of synthetic
glucocorticoids as a premedication in chemotherapy treatment, which
could potentially attenuate the effectiveness of chemotherapy
(58). EPI and NE act as the
physiological agonists for β-adrenergic receptors, with EPI
preferentially binding to β2-adrenergic receptors and NE
binding with higher affinity to β1-adrenergic receptors
(59). Owing to the function of
β-adrenergic receptors as G-protein-coupled cell membrane
receptors, the psychological stress-stimulated release of EPI and
NE triggers the formation of cAMP and activation of protein kinase
A, leading to phosphorylation of the transcription factor cAMP
response element binding protein (60). Similarly, glucocorticoids interfere
with aberrant mechanisms in cancer cells by binding to
glucocorticoid receptors, leading to nuclear translocation of the
receptor-ligand complex, its binding to glucocorticoid receptor
response elements in the promoter region of target genes and the
activation of gene transcription. The activation of stress hormone
receptors signals the stress from the extracellular environment to
the tumor cell interior, leading to the growth and metastasis of
malignant cancers (61). In the
present study, it was observed that stress hormones enhanced the
stability of Wip1 protein in HepG2 cells. Considering that no
alteration in Wip1 mRNA expression was detected, we hypothesize
that stress hormone-induced Wip1 upregulation occurs via
post-transcriptional regulation induced by the activation of
glucocorticoid or β-adrenergic receptors. However, further
assessments of protein degradation pathways following stress
hormone treatments would be helpful for determining the mechanism
by which stress hormones regulate the stability of Wip1 and protect
the protein from degradation.
It is essential to gain an improved understanding of
how cancerous cells adapt to chronic stress for the translation of
research to the clinic. In addition to transient effects such as
heart rate changes or immune cell trafficking, the increased
production of sympathetic and other adrenal hormones in response to
stress causes long-lasting consequences such as permanent DNA
damage, which results in increased cell transformation and/or
tumorigenicity. Stress hormones can induce DNA damage sensors such
as Chk1, Chk2 and E3 ubiquitin-protein ligase Mdm2, and the
resulting DNA damage can trigger cellular responses, including cell
cycle checkpoint activity, apoptosis and DNA repair pathways
(62–64). However, few studies have explored
the mechanism by which stress hormones impact cancer progression
via the induction of DNA damage. In the present research, DNA
fragmentation was observed after the treatment of human liver
cancer cells with stress hormones for 4 h and persisted after 48 h
of treatment with DEX and NE. It is noteworthy that there was no
aggravation of EPI-induced DNA damage at 48 h. The suggestion that
cells repair DNA breaks and continue to survive requires
elucidation in future studies. However, overall, adaptation
occurred, which is a process for allowing cell survival despite
persistent DNA damage. The present study further identified the
contribution of increased Wip1 protein stability to DNA damage
adaption. The importance of Wip1 is evident from the fact that it
is amplified and upregulated in liver, breast, ovarian, pancreatic,
colorectal and gastric cancers (35,65–68).
Within the DNA damage response mechanism, Wip1 acts as a
homeostatic regulator via the dephosphorylation of important DNA
damage sensor kinases, which facilitates the survival of tumor
cells following the repair of DNA damage. Adaptation is thought to
provide cancer cells with the opportunity to survive and undergo
cell division with unrepaired DNA damage (69–71).
However, the mechanism by which the accumulation of Wip1 promotes
cancer progression in response to chronic stress requires more
in-depth investigations. The relationship between Wip1 expression
and stress hormones also merits further verification in patients
with liver cancer.
Notably, the stress response is mediated by a
complex and interconnected infrastructure constituted by the
central and peripheral nervous systems, and cell-based research may
not reflect the complex process that exists in the body. The
detrimental effects of chronic stressors on tumor growth and
progression are yet to undergo systematic evaluation in
stress-based animal models. A few animal models are available for
elucidating the etiology of stress-related tumor growth and
metastasis, including the chronic restraint, social defeat and
chronic unpredictable stress models. Repetitive exposure to these
psychosocial or physical stresses mimics the features of human
pathological conditions. Moreover, chronic stress exposure can be
mimicked by treating rodents chronically with stress hormones. In
our ongoing research, the chronic restraint stress model is being
used to determine the role of Wip1 in stress-induced tumor
immunosuppression. The identification of the specific stress
hormones and receptors involved in this process are likely to
provide a fundamental understanding of the mechanisms by which the
nervous system and the tumor tissue interact. More conclusive
evidence and powerful findings could be obtained from additional
studies using different cell lines and diverse animal models in
order to minimize cell line specificity and the discrepancy between
in vitro and in vivo conditions.
In conclusion, the present study provides
preliminary evidence of the involvement of Wip1 in the tumor cell
response to stress hormones. The results provide new insights into
the mechanism underlying the effect of stress hormone signaling on
DNA damage adaption. It is anticipated that the findings may be
helpful in the development of more effective therapeutic
interventions for the prevention and treatment of liver cancer,
particularly in patients who are subjected to stress.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Jingrong Cui
(Peking University) for the gift of the human HepG2 cell line.
Funding
This study was primarily funded by the National Natural Science
Foundation of China (grant no. 81972688). Additional support was
provided by Peking University People's Hospital Scientific Research
Developmental Funds (grant no. RDGS2022-03), the Beijing Municipal
Natural Science Foundation (grant no. 7192128), the National
Science and Technology Major Projects for Major New Drugs
Innovation and Development (grant no. 2019ZX09301170) and the CAMS
Innovation Fund for Medical Sciences (CIFMS; grant no.
2021-I2M-1-026).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Conceptualization was performed by GL, WH and YL.
Research methods were carried out by GL, WH, YQ, YC, MC, XY, DK,
GW, HA and NY. Data analysis was performed by GL and WH.
Administration was performed by NY. GL wrote the original draft of
the manuscript, and WH, NY and YL reviewed and edited the
manuscript. Funding was acquired by WH, HA and YL. Supervision was
by YL. All authors read and approved the final version of the
manuscript. GL, WH and YL confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATM
|
ataxia telangiectasia mutated
|
|
ATR
|
ataxia telangiectasia and Rad3
related
|
|
Chk1
|
checkpoint kinase 1
|
|
Chk2
|
checkpoint kinase 1
|
|
DEX
|
dexamethasone
|
|
DMSO
|
dimethyl sulfoxide
|
|
EPI
|
epinephrine
|
|
FBS
|
fetal bovine serum
|
|
γ-H2AX
|
phosphorylated histone 2AX
|
|
HPA axis
|
hypothalamic-pituitary-adrenocortical
axis
|
|
NE
|
norepinephrine
|
|
PCR
|
polymerase chain reaction
|
|
PI
|
propidium iodide
|
|
SNS
|
sympathetic nervous system
|
|
Wip1
|
wild-type p53-induced phosphatase
1
|
References
|
1
|
Ebstein AM, Joseph SJ and Hernandez M:
Psychological stress and pancreatic cancer patients: A systematic
review protocol. JBI Evid Synth. 18:576–582. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abdollahi A, Panahipour H, Hosseinian S
and Allen KA: The effects of perceived stress on hope in women with
breast cancer and the role of psychological hardiness.
Psychooncology. 28:1477–1482. 2019. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharpley CF, Christie DRH, Bitsika V,
Andronicos NM, Agnew LL, Richards TM and McMillan ME: Comparing a
genetic and a psychological factor as correlates of anxiety,
depression, and chronic stress in men with prostate cancer. Support
Care Cancer. 26:3195–3200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiriac VF, Baban A and Dumitrascu DL:
Psychological stress and breast cancer incidence: A systematic
review. Clujul Med. 91:18–26. 2018.PubMed/NCBI
|
|
5
|
Zhao L, Xu J, Liang F, Li A, Zhang Y and
Sun J: Effect of chronic psychological stress on liver metastasis
of colon cancer in mice. PLoS One. 10:e01399782015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schuller HM: Effects of tobacco
constituents and psychological stress on the β-adrenergic
regulation of non-small cell lung cancer and pancreatic cancer:
Implications for intervention. Cancer Biomark. 13:133–144. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lutgendorf SK, DeGeest K, Dahmoush L,
Farley D, Penedo F, Bender D, Goodheart M, Buekers TE, Mendez L,
Krueger G, et al: Social isolation is associated with elevated
tumor norepinephrine in ovarian carcinoma patients. Brain Behav
Immun. 25:250–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shin KJ, Lee YJ, Yang YR, Park S, Suh PG,
Follo MY, Cocco L and Ryu SH: Molecular mechanisms underlying
psychological stress and cancer. Curr Pharm Des. 22:2389–2402.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elefteriou F: Chronic stress, sympathetic
activation and skeletal metastasis of breast cancer cells. Bonekey
Rep. 4:6932015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moreno-Smith M, Lutgendorf SK and Sood AK:
Impact of stress on cancer metastasis. Future Oncol. 6:1863–1881.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Romana-Souza B, Lima-Cezar GS and
Monte-Alto-Costa A: Psychological stress-induced catecholamines
accelerates cutaneous aging in mice. Mech Ageing Dev. 152:63–73.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu W, Liu S, Liang Y, Zhou Z, Bian W and
Liu X: Stress hormone cortisol enhances Bcl2 like-12 expression to
inhibit p53 in hepatocellular carcinoma cells. Dig Dis Sci.
62:3495–3500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Webster S, Chandrasekaran S, Vijayaragavan
R and Sethu G: Impact of emotional support on serum cortisol in
breast cancer patients. Indian J Palliat Care. 22:141–149. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Obradovic MMS, Hamelin B, Manevski N,
Couto JP, Sethi A, Coissieux MM, Münst S, Okamoto R, Kohler H,
Schmidt A and Bentires-Alj M: Glucocorticoids promote breast cancer
metastasis. Nature. 567:540–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Drebert Z, De Vlieghere E, Bridelance J,
De Wever O, De Bosscher K, Bracke M and Beck IM: Glucocorticoids
indirectly decrease colon cancer cell proliferation and invasion
via effects on cancer-associated fibroblasts. Exp Cell Res.
362:332–342. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pufall MA: Glucocorticoids and cancer. Adv
Exp Med Biol. 872:315–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL,
Dong XR, Zhang S, Zhang RG, Meng R, Zhu F and Wu G: Catecholamines
contribute to the neovascularization of lung cancer via
tumor-associated macrophages. Brain Behav Immun. 81:111–121. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang EV: Role for catecholamines in tumor
progression: Possible use for β-blockers in the treatment of
cancer. Cancer Biol Ther. 10:30–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Melhem-Bertrandt A, Chavez-Macgregor M,
Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD,
Hortobagyi GN and Gonzalez-Angulo AM: Beta-blocker use is
associated with improved relapse-free survival in patients with
triple-negative breast cancer. J Clin Oncol. 29:2645–2652. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Flint MS, Baum A, Chambers WH and Jenkins
FJ: Induction of DNA damage, alteration of DNA repair and
transcriptional activation by stress hormones.
Psychoneuroendocrinology. 32:470–479. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jenkins FJ, Van Houten B and Bovbjerg DH:
Effects on DNA damage and/or repair processes as biological
mechanisms linking psychological stress to cancer risk. J Appl
Biobehav Res. 19:3–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Flint MS, Baum A, Episcopo B, Knickelbein
KZ, Dougall AJ, Chambers WH and Jenkins FJ: Chronic exposure to
stress hormones promotes transformation and tumorigenicity of 3T3
mouse fibroblasts. Stress. 16:114–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Satin JR, Linden W and Phillips MJ:
Depression as a predictor of disease progression and mortality in
cancer patients: A meta-analysis. Cancer. 115:5349–5361. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Steel JL, Geller DA, Gamblin TC, Olek MC
and Carr BI: Depression, immunity, and survival in patients with
hepatobiliary carcinoma. J Clin Oncol. 25:2397–2405. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Madden KS, Szpunar MJ and Brown EB:
β-Adrenergic receptors (β-AR) regulate VEGF and IL-6 production by
divergent pathways in high β-AR-expressing breast cancer cell
lines. Breast Cancer Res Treat. 130:747–758. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lutgendorf SK, Lamkin DM, Jennings NB,
Arevalo JM, Penedo F, DeGeest K, Langley RR, Lucci JA III, Cole SW,
Lubaroff DM and Sood AK: Biobehavioral influences on matrix
metalloproteinase expression in ovarian carcinoma. Clin Cancer Res.
14:6839–6846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Zhang Y, He Z, Yin K, Li B, Zhang
L and Xu Z: Chronic stress promotes gastric cancer progression and
metastasis: An essential role for ADRB2. Cell Death Dis.
10:7882019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valente VB, de Melo Cardoso D, Kayahara
GM, Nunes GB, Tjioe KC, Biasoli ÉR, Miyahara GI, Oliveira SHP,
Mingoti GZ and Bernabé DG: Stress hormones promote DNA damage in
human oral keratinocytes. Sci Rep. 11:197012021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wani TH, Surendran S, Mishra VS,
Chaturvedi J, Chowdhury G and Chakrabarty A: Adaptation to chronic
exposure to sepantronium bromide (YM155), a prototypical survivin
suppressant is due to persistent DNA damage-response in breast
cancer cells. Oncotarget. 9:33589–33600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pechackova S, Burdova K and Macurek L:
WIP1 phosphatase as pharmacological target in cancer therapy. J Mol
Med (Berl). 95:589–599. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fiscella M, Zhang H, Fan S, Sakaguchi K,
Shen S, Mercer WE, Woude GFV, O'Connor PM and Appella E: Wip1, a
novel human protein phosphatase that is induced in response to
ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodriguez A, Naveja JJ, Torres L, de
Teresa BG, Juarez-Figueroa U, Ayala-Zambrano C, Azpeitia E, Mendoza
L and Frías S: WIP1 contributes to the adaptation of Fanconi anemia
cells to DNA damage as determined by the regulatory network of the
Fanconi anemia and checkpoint recovery pathways. Front Genet.
10:4112019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zheng R, Qu C, Zhang S, Zeng H, Sun K, Gu
X, Xia C, Yang Z, Li H, Wei W, et al: Liver cancer incidence and
mortality in China: Temporal trends and projections to 2030. Chin J
Cancer Res. 30:571–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li GB, Zhang XL, Yuan L, Jiao QQ, Liu DJ
and Liu J: Protein phosphatase magnesium-dependent 1δ (PPM1D) mRNA
expression is a prognosis marker for hepatocellular carcinoma. PLoS
One. 8:e607752013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu Z, Cao C, Xia H, Shi S, Hong L, Wei X,
Gu D, Bian J, Liu Z, Huang W, et al: Protein phosphatase
magnesium-dependent 1δ is a novel tumor marker and target in
hepatocellular carcinoma. Front Med. 10:52–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zegura B, Sedmak B and Filipic M:
Microcystin-LR induces oxidative DNA damage in human hepatoma cell
line HepG2. Toxicon. 41:41–48. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang W, Liu Y, Wang J, Yuan X, Jin HW,
Zhang LR, Zhang JT, Liu ZM and Cui JR: Small-molecule compounds
targeting the STAT3 DNA-binding domain suppress survival of
cisplatin-resistant human ovarian cancer cells by inducing
apoptosis. Eur J Med Chem. 157:887–897. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang W, Yuan X, Sun T, Fan S, Wang J,
Zhou Q, Guo W, Ran F, Ge Z, Yang H, et al: Proteasome inhibitor
YSY01A abrogates constitutive STAT3 signaling via down-regulation
of Gp130 and JAK2 in human A549 lung cancer cells. Front Pharmacol.
8:4762017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kao SH, Wang WL, Chen CY, Chang YL, Wu YY,
Wang YT, Wang SP, Nesvizhskii AI, Chen YJ, Hong TM and Yang PC:
Analysis of protein stability by the cycloheximide chase assay. Bio
Protoc. 5:e13742015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang W, Dong Z, Wang F, Peng H, Liu JY
and Zhang JT: A small molecule compound targeting STAT3 DNA-binding
domain inhibits cancer cell proliferation, migration, and invasion.
ACS Chem Biol. 9:1188–1196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang W, Dong Z, Chen Y, Wang F, Wang CJ,
Peng H, He Y, Hangoc G, Pollok K, Sandusky G, et al: Small-molecule
inhibitors targeting the DNA-binding domain of STAT3 suppress tumor
growth, metastasis and STAT3 target gene expression in vivo.
Oncogene. 35:783–792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang W, Zhou Q, Yuan X, Ge ZM, Ran FX,
Yang HY, Qiang GL, Li RT and Cui JR: Proteasome inhibitor YSY01A
enhances cisplatin cytotoxicity in cisplatin-resistant human
ovarian cancer cells. J Cancer. 7:1133–1141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tapryal N, Vivek GV and Mukhopadhyay CK:
Catecholamine stress hormones regulate cellular iron homeostasis by
a posttranscriptional mechanism mediated by iron regulatory
protein: Implication in energy homeostasis. J Biol Chem.
290:7634–7646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ma J, Xue M, Zhang S, Cheng L, Qian W,
Duan W and Shen X: Resveratrol inhibits the growth of tumor cells
under chronic stress via the ADRB-2-HIF-1α axis. Oncol Rep.
41:1051–1058. 2019.PubMed/NCBI
|
|
47
|
Cayla C, Schaak S, Roquelaine C, Gales C,
Quinchon F and Paris H: Homologous regulation of the
α2C-adrenoceptor subtype in human hepatocarcinoma, HepG2. Br J
Pharmacol. 126:69–78. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Uen YH, Ko PH, Yin PH, Liu TY, Chi CW and
Lui WY: Glucocorticoid protects hepatoma cells against metabolic
stress-induced cell death. Int J Oncol. 33:1263–1270.
2008.PubMed/NCBI
|
|
49
|
Li M, Chen F, Liu CP, Li DM, Li X, Wang C
and Li JC: Dexamethasone enhances trichosanthin-induced apoptosis
in the HepG2 hepatoma cell line. Life Sci. 86:10–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Oliver AW, Paul A, Boxall KJ, Barrie SE,
Aherne GW, Garrett MD, Mittnacht S and Pearl LH: Trans-activation
of the DNA-damage signalling protein kinase Chk2 by T-loop
exchange. EMBO J. 25:3179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2, and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shi T, van Soest DMK, Polderman PE,
Burgering BMT and Dansen TB: DNA damage and oxidizing conditions
activate p53 through differential upstream signaling pathways. Free
Radic Biol Med. 172:298–311. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ozaki T and Nakagawara A: Role of p53 in
cell death and human cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang XP, Liu F and Wang W: Two-phase
dynamics of p53 in the DNA damage response. Proc Natl Acad Sci USA.
108:8990–8995. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ogunwobi OO, Harricharran T, Huaman J,
Galuza A, Odumuwagun O, Tan Y, Ma GX and Nguyen MT: Mechanisms of
hepatocellular carcinoma progression. World J Gastroenterol.
25:2279–2293. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sood AK, Bhatty R, Kamat AA, Landen CN,
Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S and Cole SW:
Stress hormone-mediated invasion of ovarian cancer cells. Clin
Cancer Res. 12:369–375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Reeder A, Attar M, Nazario L, Bathula C,
Zhang A, Hochbaum D, Roy E, Cooper KL, Oesterreich S, Davidson NE,
et al: Stress hormones reduce the efficacy of paclitaxel in triple
negative breast cancer through induction of DNA damage. Br J
Cancer. 112:1461–1470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Alyusuf R, Wazir JF, Brahmi UP, Fakhro AR
and Bakhiet M: The immunoexpression of glucocorticoid receptors in
breast carcinomas, lactational change, and normal breast epithelium
and its possible role in mammary carcinogenesis. Int J Breast
Cancer. 2017:14030542017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Maki T, Kontula K and Harkonen M: The
β-adrenergic system in man: Physiological and pathophysiological
response. Regulation of receptor density and functioning. Scand J
Clin Lab Invest Suppl. 201:25–43. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lorton D and Bellinger DL: Molecular
mechanisms underlying β-adrenergic receptor-mediated cross-talk
between sympathetic neurons and immune cells. Int J Mol Sci.
16:5635–5665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Escoter-Torres L, Caratti G, Mechtidou A,
Tuckermann J, Uhlenhaut NH and Vettorazzi S: Fighting the fire:
Mechanisms of inflammatory gene regulation by the glucocorticoid
receptor. Front Immunol. 10:18592019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lindström MS, Bartek J and Maya-Mendoza A:
p53 at the crossroad of DNA replication and ribosome biogenesis
stress pathways. Cell Death Differ. 29:972–982. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Molinaro C, Martoriati A and Cailliau K:
Proteins from the DNA damage response: Regulation, dysfunction, and
anticancer strategies. Cancers (Basel). 13:38192021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Weitzman MD and Weitzman JB: What's the
Damage? The impact of pathogens on pathways that maintain host
genome integrity. Cell Host Microbe. 15:283–294. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lambros MB, Natrajan R, Geyer FC,
Lopez-Garcia MA, Dedes KJ, Savage K, Lacroix-Triki M, Jones RL,
Lord CJ, Linardopoulos S, et al: PPM1D gene amplification and
overexpression in breast cancer: a qRT-PCR and chromogenic in situ
hybridization study. Mod Pathol. 23:1334–1345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Peng TS, He YH, Nie T, Hu XD, Lu HY, Yi J,
Shuai YF and Luo M: PPM1D is a prognostic marker and therapeutic
target in colorectal cancer. Exp Ther Med. 8:430–434. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sun GG, Wang YD, Liu Q and Hu WN:
Expression of Wip1 in kidney carcinoma and its correlation with
tumor metastasis and clinical significance. Pathol Oncol Res.
21:219–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fuku T, Semba S, Yutori H and Yokozaki H:
Increased wild-type p53-induced phosphatase 1 (Wip1 or PPM1D)
expression correlated with downregulation of checkpoint kinase 2 in
human gastric carcinoma. Pathol Int. 57:566–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Carr MI and Jones SN: Regulation of the
Mdm2-p53 signaling axis in the DNA damage response and
tumorigenesis. Transl Cancer Res. 5:707–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Feroz W and Sheikh AMA: Exploring the
multiple roles of guardian of the genome: P53. Egypt J Med Hum
Genet. 21:492020. View Article : Google Scholar
|
|
71
|
Choi DW, Na W, Kabir MH, Yi E, Kwon S,
Yeom J, Ahn JW, Choi HH, Lee Y, Seo KW, et al: WIP1, a homeostatic
regulator of the DNA damage response, is targeted by HIPK2 for
phosphorylation and degradation. Mol Cell. 51:374–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|