Introduction
Breast cancer (BC) is one of the most common primary
malignant tumors among women worldwide and is a serious threat to
women's health (1). In recent
decades, the incidence of BC has rapidly increased, with ≥1 million
cases newly diagnosed each year (2). Triple-negative BC (TNBC) is the most
aggressive subtype of BC, and is defined as a lack of expression of
estrogen receptor (ER), progesterone receptor (PR) and human
epidermal growth factor receptor 2 (HER2) (3,4). At
present, systemic chemotherapy is the main treatment for patients
with TNBC (5). TNBC is more
aggressive and prone to local recurrence and lymph node metastasis
compared with other types of BC, leading to poor prognosis or
clinical outcomes. In Chinese female patients, the 5-year survival
rate of early-stage TNBC is 77%, which is much lower than that for
other subsets of early-stage BC (6). Therefore, it is necessary to identify
new alternatives for TNBC treatment.
MYC proto-oncogene (MYC) is a protein-coding
transcription factor, and its family contains B-MYC, C-MYC, MYCL
proto-oncogene (MYCL), N-MYC and S-MYC (7). In human primary small cell lung
cancer (SCLC), MYCL was first reported to be amplified, which is
usually accompanied by overexpression (8). Previous studies have shown that MYC
is dysregulated in numerous types of cancer, including lung cancer,
Burkitt's lymphoma, colon cancer and BC (8–11).
In cancer, MYC has synergistic effects with other transcription
factors and target genes to regulate numerous life processes,
including cell growth, apoptosis, cell cycle and tumorigenesis
(12,13). In addition, MYCL has been reported
to accelerate proliferation and increase metastatic dissemination
of SCLC (14,15). Previous studies have also revealed
that MYC expression is dysregulated in 30–50% of cases of
high-grade BC. Compared with in normal breast tissues, MYC
expression in tumor tissues has been shown to be decreased, and
among the major subclasses, MYC expression is relatively high in
TNBC (16). Compared with C-MYC or
N-MYC, which drive several types of human cancer (17–20),
there are fewer studies on MYCL. Furthermore, to the best of our
knowledge, no previous study has reported the genetic changes of
the MYCL gene related to TNBC.
The JAK/STAT pathway is considered to be an
evolutionarily conserved signaling pathway, which is regulated by a
variety of interferons, cytokines, related molecules and growth
factors (21). It is well known
that the JAK/STAT pathway serves a key role in a variety of
biological processes, including cell differentiation, apoptosis,
proliferation, survival and immune response (22). It has previously been reported that
the JAK/STAT3 autocrine activation loop is a vital driver of
metastasis and progression of BC (23). However, it is unclear as to whether
the JAK/STAT3 pathway can be regulated by MYCL in TNBC. The present
study explored the role of MYCL in TNBC progression, and further
explored the potential regulatory mechanism of MYCL in activating
the JAK/STAT3 pathway.
Materials and methods
Cell lines and cell culture
The normal human breast epithelial cell line
(MCF-10A), the 293 cell line and four TNBC cell lines (HCC1143,
MDA-MB-231, BT-549 and MDA-MB-453) were acquired from American Type
Culture Collection. Cells were incubated in Dulbecco's modified
Eagle's medium containing 10% fetal bovine serum (FBS), and 1%
penicillin and streptomycin (all from Thermo Fisher Scientific,
Inc.). The cells were fostered at 37°C in an atmosphere containing
5% CO2 and the medium was changed every 2–3 days.
Cell transfection
MYCL small interfering RNA (siRNAs) (si-MYCL#1,
5′-GACTACGACTCGTACCAGCACTATT-3′; si-MYCL#2,
5′-CAGCACTATTTCTACGACTATGACT-3′; and si-MYCL#3,
5′-CAAGCGACTCGGAGAATGAAGAAAT-3′) and negative control (si-NC,
5′-TTCTCCGAACGTGTCACGT-3′) were purchased from the Shanghai
GenePharma Co., Ltd. The MYCL pcDNA3.1 overexpression vector
(pc-MYCL) and negative control (pc-NC) were synthesized by Sangon
Biotech Co., Ltd. The human TNBC cells (1×105) were
seeded and cultured in 24-well plates for 1 day. After 72 h of cell
culture, si-MYCL or si-NC (50 nM)was used to transfect MDA-MB-453
cells for 96 h at 37°C using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) to construct a
MYCL-silenced cell model. In addition, MDA-MB-231 cells were
transfected with pc-MYCL or pc-NC (2 µg) using
Lipofectamine® 2000 for 96 h at 37°C to construct a
MYCL-overexpressed cell model. Subsequent experiments were
performed after 48 h of transfection. Furthermore, to confirm the
effect of JAK on MYCL expression, a JAK inhibitor, 1 µM ruxolitinib
(Ruxo; Shanghai YuanyeBio-Technology Co., Ltd.), was added into the
medium to treat MDA-MB-231 cells transfected with pc-MYCL or pc-NC
for 24 h at 37°C.
Cell apoptosis analysis
The Annexin V-PE/7-AAD Apoptosis Detection Kit
(Vazyme Biotech Co., Ltd.) was used to examine the apoptosis of
MDA-MB-453 and MDA-MB-231 cells. Briefly, cells (1×105
cells/well in 6-well plates) were suspended in 100 µl binding
buffer and incubated with 5 µl Annexin V-PE and 5 µl 7-AAD for 10
min in the dark at room temperature. Within 1 h of staining, flow
cytometry (FACScan; BD Biosciences) was performed to evaluate the
apoptotic rate of TNBC cells. The apoptotic rate was calculated as
the percentage of early and late apoptotic cells.
Colony formation assay
After 48 h of transfection, MDA-MB-231 or MDA-MB-453
cells (1×103 cells/well) were transferred to 6-well
plates and routinely cultured for 14 days to form obvious colonies.
Subsequently, the colonies (>50 cells) were fixed in 4%
formaldehyde at 37°C for 20 min, followed by staining with 1%
crystal violet (Sangon Biotech Co., Ltd.) for 15 min. Finally,
images of the cell colonies were captured using a digital Sight
camera (Nikon Corp.) and counted from five random fields under a
light microscope (Olympus Corp.).
Wound healing assay
Briefly, after 48 h of transfection, MDA-MB-231 or
MDA-MB-453 cells (50 µl; 5×105 cells/ml) were inoculated
into 6-cm culture plates with RPMI-1640 medium (Thermo Fisher
Scientific, Inc.) containing 10% FBS. Once the cells reached 90%
confluence, a single linear scratch was drawn in the middle of the
cell monolayer with a 200-µl pipette tip. Subsequently, the cell
debris was removed by flushing three times with PBS. The cells were
then cultured in RPMI-1640 medium supplemented with 1% FBS for 48
h. Finally, images of the wounds were captured by a light
microscope (Olympus Corp.). The relative migration rate was
calculated as follows: Relative migration rate (%)=(wound distance
at 0 h-wound distance at 48 h)/wound distance at 0 h ×100.
Transwell assay
The Transwell assay was performed to evaluate cell
invasion in vitro. The Transwell chambers (pore size, 8 µm) were
coated with Matrigel (BD Biosciences) and incubated for 1 h at
37°C. TNBC cells (5×104 cells/well in a 24-well plate)
were suspended in RPMI-1640 serum-free medium on the upper surface
of the chambers. Meanwhile, 500 µl RPMI-1640 medium with 10% FBS
was added to the bottom chamber. After the cells were cultured at
37°C (5% CO2) for 1 day, cells on the upper side of the
membrane were removed using a cotton swab, followed by washing with
PBS three times. Subsequently, the cells on the opposite side of
the membrane were fixed with 4% paraformaldehyde at room
temperature for 20 min and stained with 0.2% crystal violet for 30
min at indoor temperature. The images were finally captured under a
light microscope (Nikon Corp.). Five visual fields were randomly
selected and the average number of stained cells was counted.
Reverse transcription-quantitative PCR
(RT-qPCR)
TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was used to extract total RNA from TNBC
cells. According to the manufacturer's protocol, total RNA (500 ng)
was reverse transcribed to cDNA using the PrimeScript RT reagent
Kit Perfect Real-Time kit (Takara Bio, Inc.). qPCR was performed to
detect the mRNA expression levels of MYCL using SYBR Master Mixture
(Takara Bio, Inc.) on a 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) as follows:
Pre-denaturation at 95°C for 2 min; 45 cycles of denaturation at
95°C for 5 sec, annealing at 55°C for 15 sec and extension at 68°C
for 30 sec; and finally, a melting curve analysis was performed
from 65 to 95°C for 3 min. The final relative expression levels of
MYCL were calculated using the 2−ΔΔCq method (24) and normalized to GAPDH. The PCR
primers of MYCL and GAPDH were as follows: MYCL, forward
5′-CCAAGCGACTCGGAGAATGA-3′ and reverse 5′-TTGGGAGCAGCTTTCTGGAG-3′;
GAPDH, forward 5′-CATGTTGCAACCGGGAAGGA-3′ and reverse
5′-CGCCCAATACGACCAAATCAG-3′.
Western blotting
Total protein was extracted from cells and xenograft
tumor tissues using RIPA lysis buffer with protease inhibitors
(Roche Diagnostics) for 30 min on ice. The concentration of all
proteins extracted from the supernatants of cell lysates was
determined using a Protein Assay kit (BCA; Takara Bio, Inc.).
Subsequently, 40 µg protein was separated by SDS-PAGE on 12% gels
and transferred to PVDF membranes (MilliporeSigma). The membranes
were blocked with 5% non-fat dry milk dissolved in PBS −0.1% Tween
for 1 h at room temperature, and primary antibodies against MYCL
(cat. no. SAB1410807; 1:1,000), E-cadherin (cat. no. SAB5700789;
1:1,000), N-cadherin (cat. no. SAB5700641; 1:1,000), Vimentin (cat.
no. SAB1305096; 1:1,000), phosphorylated (p)-JAK1 (cat. no.
SAB4504446; 1:1,000), JAK1 (cat. no. SAB4300393; 1:1,000), p-JAK2
(cat. no. SAB4300124; 1:1,000), JAK2 (cat. no. SAB4501601;
1:1,000), p-STAT3 (cat. no. SAB5700362; 1:1,000), STAT3 (cat. no.
SAB5700069; 1:1,000), C-MYC (cat. no. SAB5700727; 1:1,000), Cyclin
D1 (cat. no. SAB4502602; 1:1,000), Bcl-2 (cat. no. SAB4500003;
1:1,000) and GAPDH (cat. no. ABS16; 1:1,000) (all from
MilliporeSigma) overnight at 4°C. After washing three times using
TBS −0.1% Tween, the membranes were incubated with a HRP-conjugated
secondary antibody (cat. no. A16096; 1:10,000; Thermo Fisher
Scientific, Inc.) for 2 h at 25°C. The protein expression intensity
was determined using an enhanced chemiluminescence reagent (Thermo
Fisher Scientific, Inc.).
Bioinformatics analysis
The UALCAN database (http://ualcan.path.uab.edu) based on The Cancer Genome
Atlas (TCGA) database was used to observe the mRNA expression of
MYCL across various types of cancer and different breast cancer
subtypes. The Gene Expression Profiling Interactive Analysis
(GEPIA) database (http://gepia.cancer-pku.cn/index.html) was used to
analyze MYCL expression in breast invasive carcinoma (BRCA). The
Gene Expression Omnibus (GEO) TNBC GSE45498 dataset (25) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45498)
was downloaded to analyze the MYCL mRNA expression in normal and
TNBC tumor tissues using GraphPad Prism 7.0 (GraphPad Software,
Inc.). The potential effect of MYCL on overall survival (OS) and
disease-free survival (DFS) of patients with TNBC was analyzed
using the Kaplan-Meier plotter (http://kmplot.com/analysis/). Gene Set Enrichment
Analysis (GSEA) was conducted on the GSE45498 dataset using the R
package clusterProfiler (26) to
explore MYCL expression and predict the potential signaling
pathway.
Lentiviral infection
MYCL short hairpin RNA (sh-MYCL) and a negative
control (sh-NC) were packaged into the pGCSIL-GFP lentiviral
vector, which was purchased from Shanghai GenePharma Co., Ltd.
Lentivirus packaging was performed using the second-generation
lentivirus packaging kit (Shanghai GeneChem Co., Ltd.) at 37°C for
15 min. For lentivirus packaging, 10 µg lentiviral plasmid
pGCSIL-GFP and two helper plasmids (5 µg pHelper 1 and 5 µg pHelper
2) were incubated with Lenti-Easy Packaging Mix (1 ml) at 37°C for
15 min. Subsequently, the mixture was incubated for another 20 min
in Lipofectamine 2000 and applied to 293T cells for transfection.
The cells were transfected with lentiviruses at 37°C for 6 h, the
medium was changed and the viral particles were collected after 3
days. The transfected cells were filtered using a 0.45-µM mesh and
were concentrated by ultracentrifugation at 70,000 × g at 4°C for 2
h. The supernatant was collected for detecting viral titers. The
MDA-MB-453 cell line was cultured to >80% confluence and was
cultured with diluted lentiviruses at a multiplicity of infection
of 10 and with polybrene (MilliporeSigma) for 24 h at 37°C.
Subsequently, fresh culture medium was used to replace the spent
medium and GFP-labeled cells with a lentivirus transduction rate
>80% at 72 h were screened out; stable expressing clones were
selected using 4 µg/ml puromycin. The transduction efficacy of
sh-MYCL was verified by western blotting.
Xenograft tumor assay
The experimental protocol of the present study was
performed in accordance with the Guide for the Care and Use of
Laboratory Animals (27) and was
approved by the Second Hospital of Shanxi Medical University
(approval no. SXDW20210512; Taiyuan, China). Eight 6-week-old
female BALB/c nude mice (weight, 20±2 g) were obtained from Jinan
Pengyue Experimental Animal Breeding Co., Ltd. All mice were housed
in a specific pathogen-free animal facility with free access to
water and food at 22±1°C with 55±2% humidity and a 12-h light/dark
cycle. The mice were randomly divided into two groups (n=4/group).
MDA-MB-453 cells (5×106 cells) stably transduced with
sh-MYCL or sh-NC were subcutaneously injected into the back of
mice. The length and width of xenograft tumors were measured
weekly. The volumes were calculated on the basis of the formula V
(mm3)=length × width2/2. After 5 weeks, the
mice were sacrificed via an intravenous injection of excess
pentobarbital sodium (100 mg/kg), and the xenograft tumors were
obtained, imaged and weighed. The protein expression levels of MYCL
and JAK/STAT3 pathway-related proteins were detected using western
blotting.
Statistical analysis
All in vitro experiments were carried out in
triplicate. All data were analyzed using SPSS 20.0 (IBM Corp.) and
are presented as the mean ± SD. The significant differences between
two groups were determined by unpaired Student's t-test. The
significant differences among multiple groups were determined by
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
MYCL is upregulated in TNBC
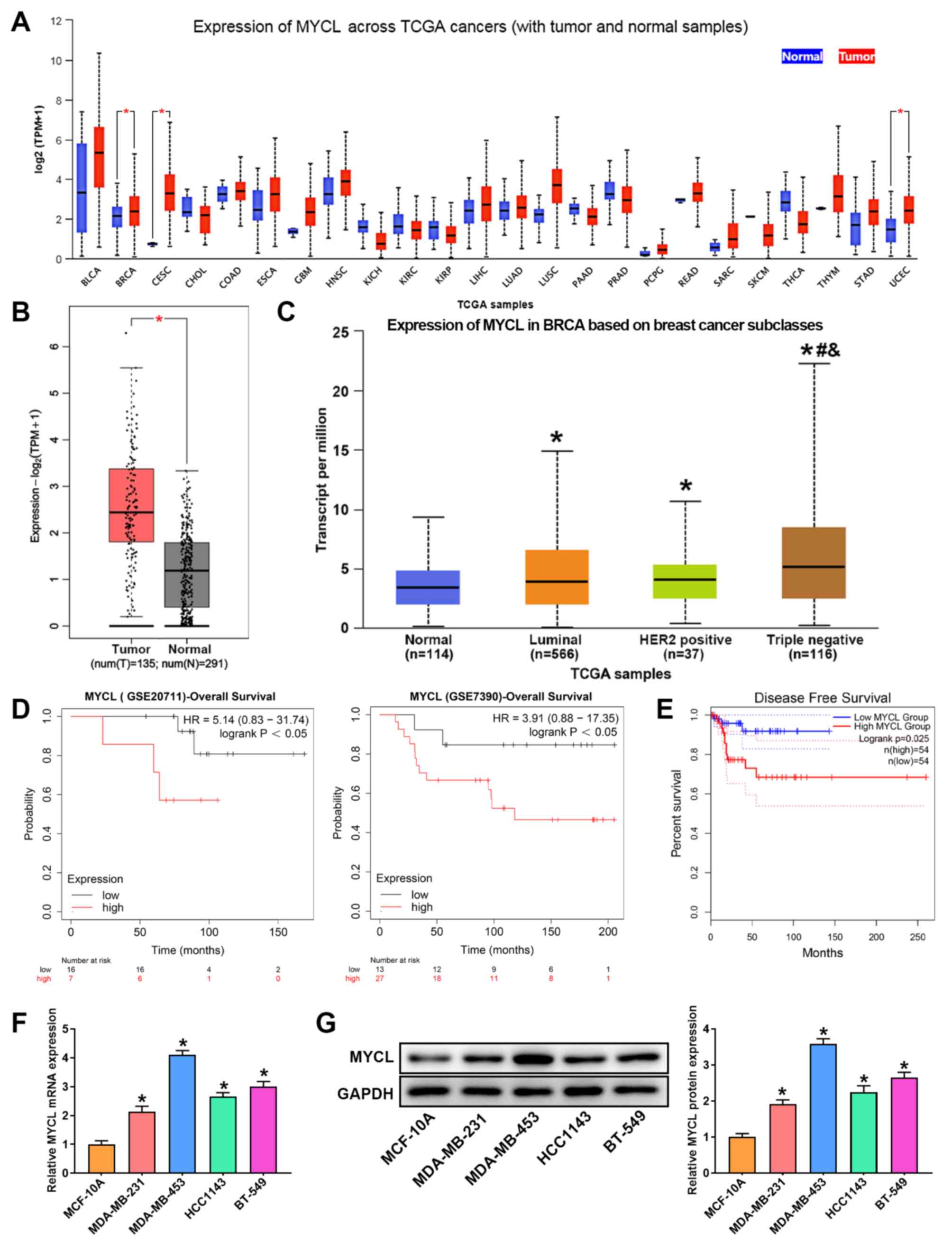
The results of the pan-cancer analysis are shown in
Fig. 1A, MYCL was revealed to be
significantly upregulated in BRCA, cervical squamous cell carcinoma
and uterine corpus endometrial carcinoma. Subsequently, the GEPIA
was used to analyze MYCL expression in BRCA, and the results
demonstrated that MYCL expression was significantly increased in
tumor compared with in normal samples (Fig. 1B). To fully understand the
expression of MYCL in patients with BC, MYCL expression based on
major subclasses was analyzed using the UALCAN tool. As shown in
Fig. 1C, the mRNA expression
levels of MYCL were significantly upregulated in different subtypes
of BC compared with in normal samples, and they were particularly
higher in TNBC (Fig. 1C).
Additionally, the Kaplan-Meier plotter indicated that compared with
patients with low MYCL expression, patients with TNBC and high MYCL
expression had poorer OS and DFS (Fig.
1D and E). Subsequently, MYCL expression was detected in
several TNBC cell lines and in a normal human breast epithelial
cell line by RT-qPCR and western blotting. Compared with in the
MCF-10A normal breast epithelial cell line, the expression levels
of MYCL in TNBC cells were significantly upregulated, but not in a
consistent manner. Among them, MYCL expression was the lowest in
MDA-MB-231 cells and was the highest in MDA-MB-453 cells (Fig. 1F and G). These findings indicated
that abnormal expression of MYCL may be related to TNBC
progression.
MYCL silencing induces the apoptosis
and suppresses the proliferation of TNBC cells
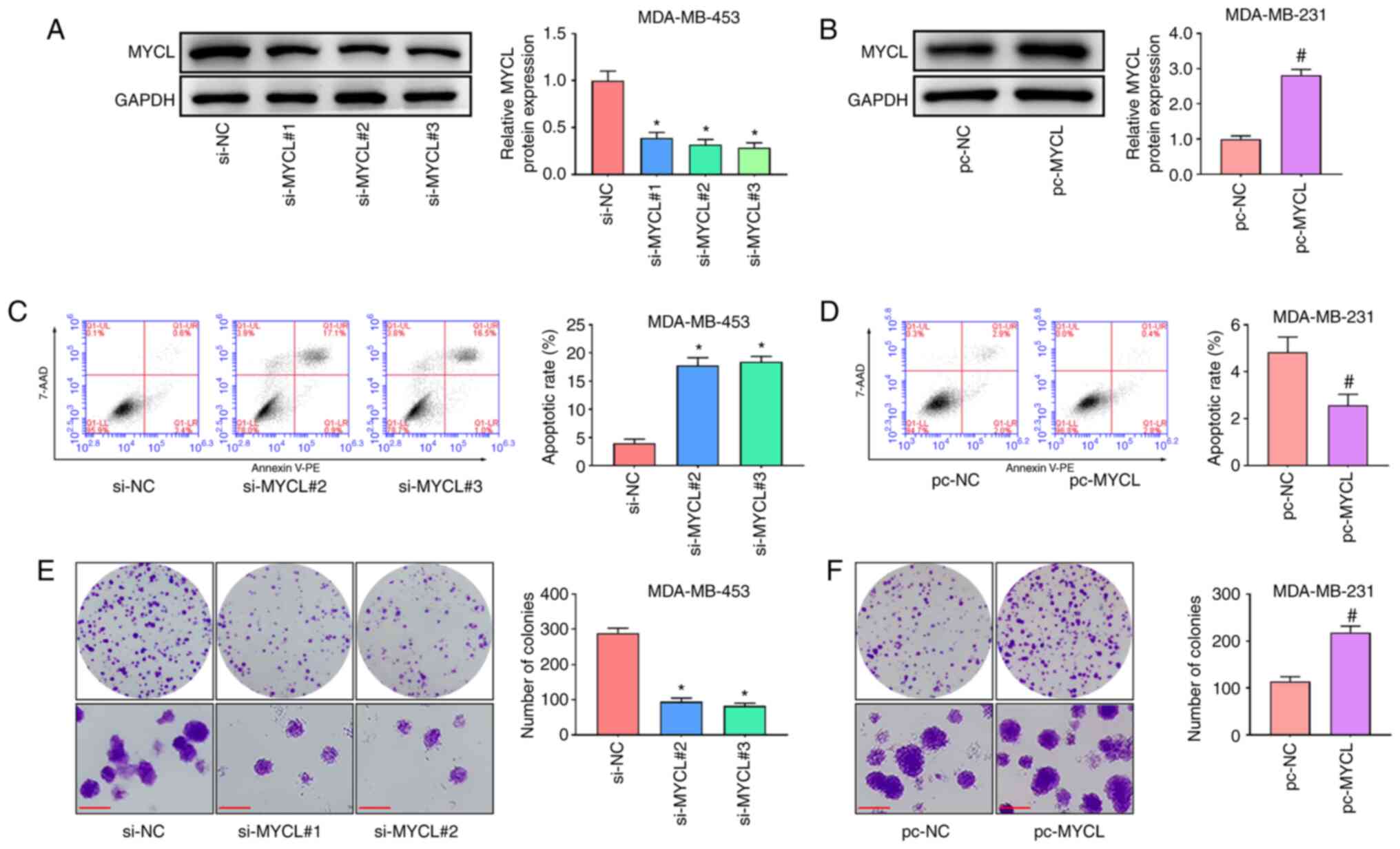
To further investigate the role of MYCL in TNBC
progression, MDA-MB-453 and MDA-MB-231 cells were selected to
perform in vitro analyses. Prior to functional experiments, the
knockdown efficiencies of three siRNAs targeting the back-splicing
sites of MYCL were verified in MDA-MB-453 cells and the efficiency
of the MYCL overexpression vector in MDA-MB-231 cells was assessed
using western blotting. MYCL expression was significantly reduced
in all three si-MYCL groups, particularly in the si-MYCL#2 and
si-MYCL#3 groups, compared with that in the si-NC group (Fig. 2A); therefore, these siRNAs were
chosen for functional experiments. Furthermore, MYCL expression was
significantly increased in the pc-MYCL group compared with that in
the pc-NC group (Fig. 2B). Cell
apoptosis and proliferation were subsequently detected using flow
cytometry and colony formation assays, respectively. The results
demonstrated that MYCL silencing induced the apoptosis and
inhibited the proliferation of MDA-MB-453 cells compared with that
in the cells transfected with si-NC (Fig. 2C and E). Conversely, MYCL
overexpression suppressed the apoptosis and promoted the
proliferation of MDA-MB-231 cells compared with that in the pc-NC
group (Fig. 2D and F). These
results indicated that MYCL silencing induced the apoptosis and
suppressed the proliferation of TNBC cells.
MYCL silencing suppresses the
migration and invasion of TNBC cells
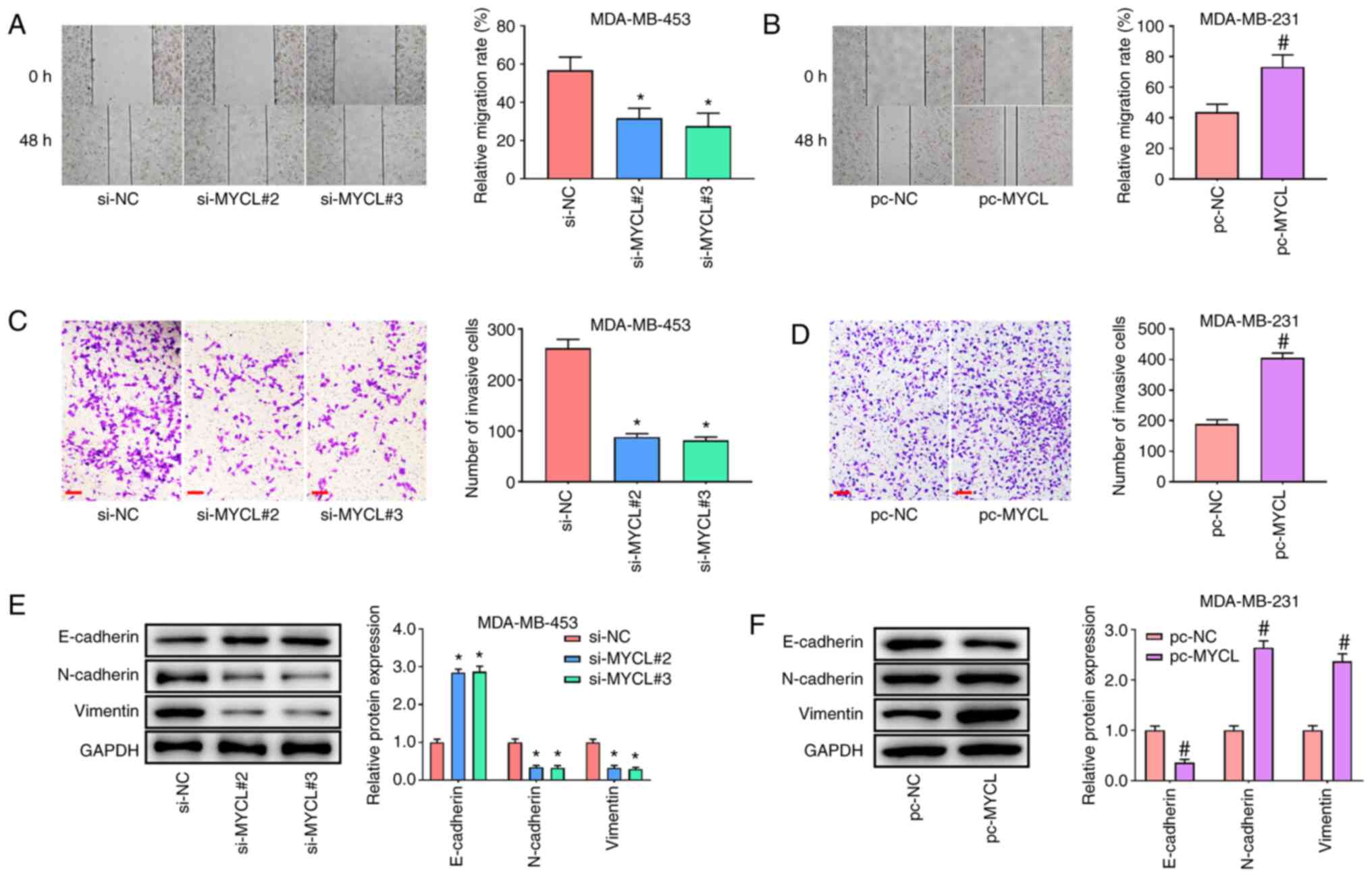
To further explore whether MYCL knockdown or
overexpression affected TNBC cell migration and invasion, further
experiments were performed. The wound healing assay indicated that
MYCL silencing significantly impaired the migratory ability of
MDA-MB-453 cells in both si-MYCL#1 and si-MYCL#2 groups compared
with that in the si-NC group (Fig.
3A). By contrast, MYCL overexpression had the opposite effect
on MDA-MB-231 cell migration (Fig.
3B). Furthermore, the Transwell assay indicated that MYCL
silencing significantly decreased the number of invasive MDA-MB-453
cells compared with that in the si-NC group (Fig. 3C), whereas the opposite effect was
observed when MYCL was overexpressed in MDA-MB-231 cells (Fig. 3D). Furthermore, as expected,
compared with those in the si-NC group, silencing of MYCL
significantly promoted the expression levels of E-cadherin, and
inhibited those of Vimentin and N-cadherin, in MDA-MB-453 cells
(Fig. 3E). Conversely, MYCL
overexpression reduced the expression levels of E-cadherin, and
enhanced those of Vimentin and N-cadherin, in MDA-MB-231 cells
compared with those in the pc-NC group (Fig. 3F). These findings provided strong
evidence that the silencing of MYCL suppressed the tumorigenic
potential of TNBC cells.
MYCL activates the JAK/STAT3
pathway
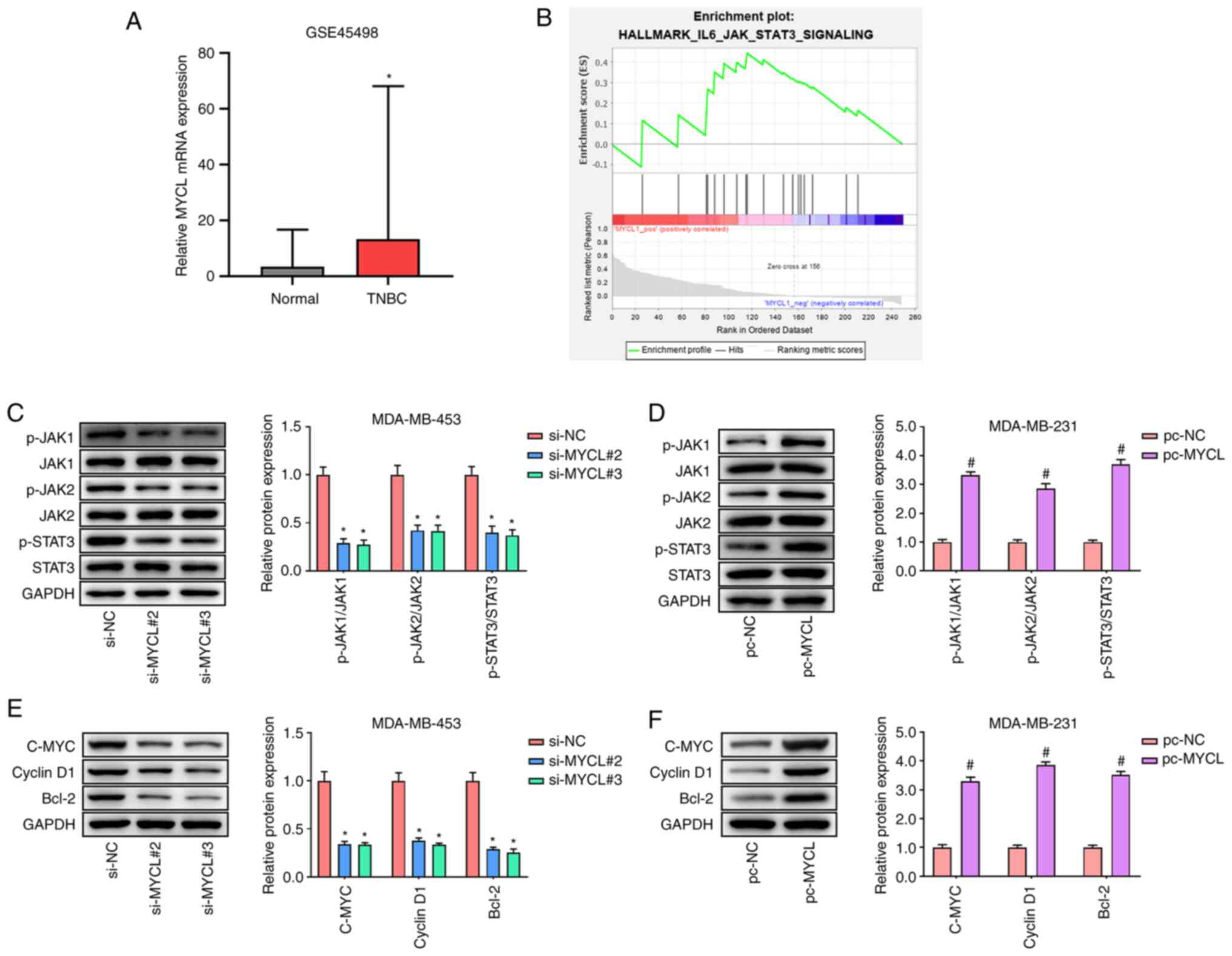
To investigate the underlying mechanisms of MYCL in
TNBC, the GSE45498 TNBC dataset was used to predict the signaling
pathway related to MYCL through GSEA. GSE45498 dataset analysis
showed that MYCL mRNA expression was significantly upregulated in
TNBC tissues compared with that in normal tissues (Fig. 4A). Furthermore, the GSEA showed
that MYCL activated the JAK/STAT3 pathway (Fig. 4B). The proteins involved in the
JAK/STAT3 pathway were subsequently evaluated by western blotting.
The results indicated that silencing of MYCL significantly reduced
the expression levels of p-JAK1/JAK1, p-JAK2/JAK2 and p-STAT3/STAT3
in MDA-MB-453 cells compared with in the si-NC group (Fig. 4C), whereas MYCL overexpression
significantly increased the expression levels of these proteins
compared with in the pc-NC group (Fig.
4D). Additionally, the protein expression levels of STAT3
downstream proteins, C-MYC, Cyclin D1 and Bcl-2, were also detected
by western blotting. MYCL silencing significantly reduced the
expression levels of C-MYC, Cyclin D1 and Bcl-2 in MDA-MB-453 cells
compared with in the si-NC group (Fig.
4E), whereas MYCL overexpression significantly enhanced the
expression levels of these proteins compared with in the pc-NC
group (Fig. 4F). These findings
demonstrated that MYCL could activate the JAK/STAT3 pathway in
TBNC.
Inhibition of the JAK/STAT3 pathway
eliminates the effect of MYCL on TNBC cells
To confirm the association between MYCL and the
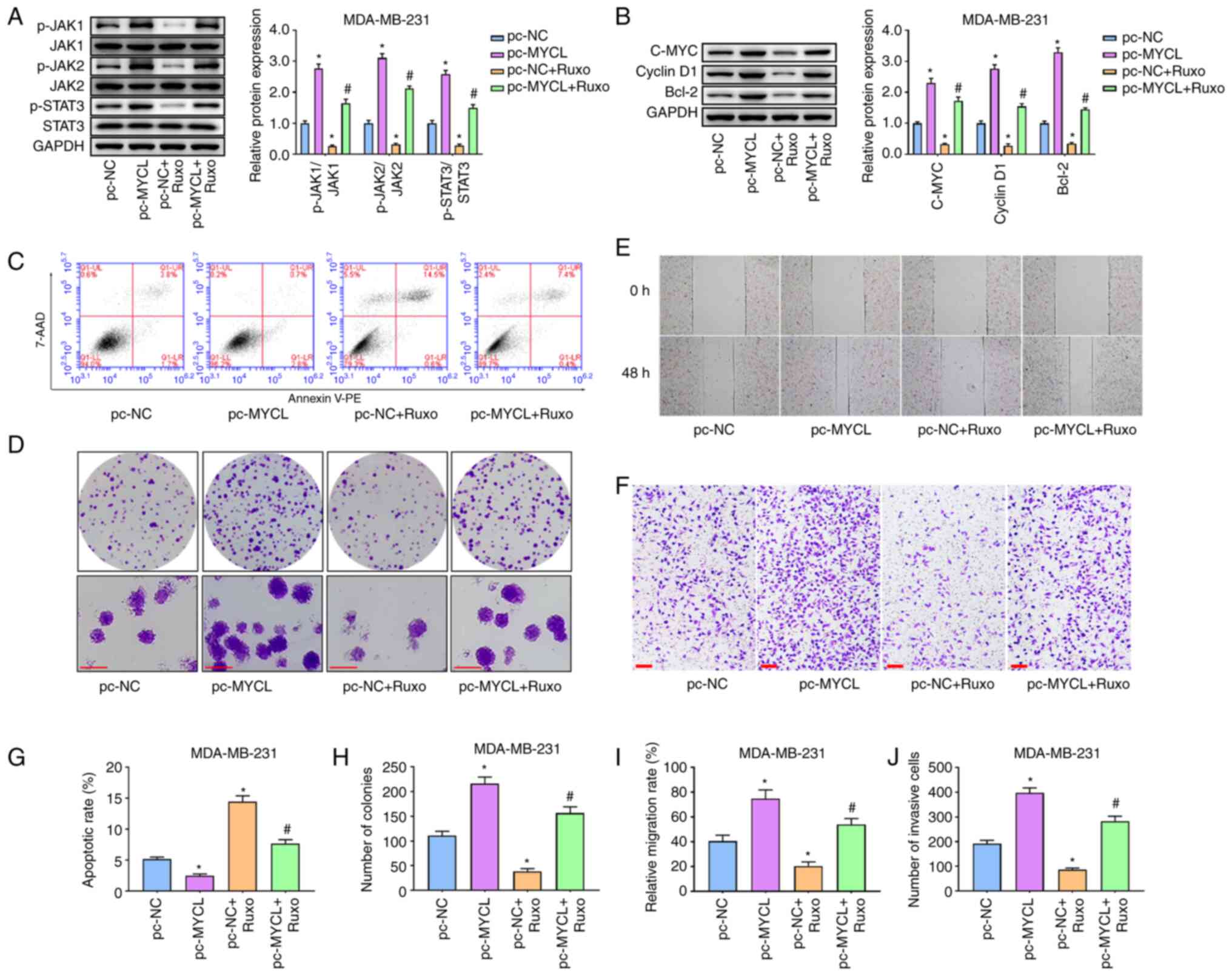
JAK/STAT3 pathway in TNBC development, Ruxo, a JAK inhibitor, was
used to treat the transfected TNBC cells. Initially, western
blotting revealed that compared with in the pc-MYCL group, the
expression levels of p-JAK1/JAK1, p-JAK2/JAK2 and p-STAT3/STAT3 in
the JAK/STAT3 pathway were significantly inhibited in the pc-MYCL +
Ruxo group, indicating that inhibition of JAK could weaken the
effect of MYCL overexpression on JAK/STAT3 signaling pathway in
TNBC cells (Fig. 5A). Similar
results were obtained in the detection of C-MYC, Cyclin D1 and
Bcl-2 (Fig. 5B). Subsequently,
experiments were performed to confirm whether MYCL affected the
biological behaviors of TNBC cells through the JAK/STAT3 pathway.
Flow cytometry revealed that MYCL overexpression significantly
inhibited apoptosis, whereas Ruxo significantly enhanced the
apoptosis of MDA-MB-231 cells compared with in the pc-NC group.
Notably, the inhibitory effect of MYCL overexpression on apoptosis
was significantly reversed by the JAK inhibitor Ruxo. The apoptosis
of MDA-MB-231 cells was significantly increased in the pc-MYCL +
Ruxo group compared with that in the pc-MYCL group (Fig. 5C and G). The colony formation assay
revealed that MYCL overexpression enhanced cell proliferation,
whereas Ruxo inhibited the proliferation of MDA-MB-231 cells
compared with in the pc-MYCL group. Furthermore, Ruxo reversed the
effect of MYCL overexpression on the proliferative ability of
MDA-MB-231 cells (Fig. 5D and H).
Consistent with the aforementioned results, MYCL overexpression
significantly promoted the migration and invasion of MDA-MB-231
cells compared with that in the pc-NC group, whereas Ruxo
significantly suppressed the effect of MYCL overexpression on the
migration and invasion of TBNC cells (Fig. 5E, F, I and J). Taken together, MYCL
may affect the biological behaviors of TNBC cells by activating the
JAK/STAT3 pathway.
MYCL silencing inhibits TNBC tumor
growth in vivo
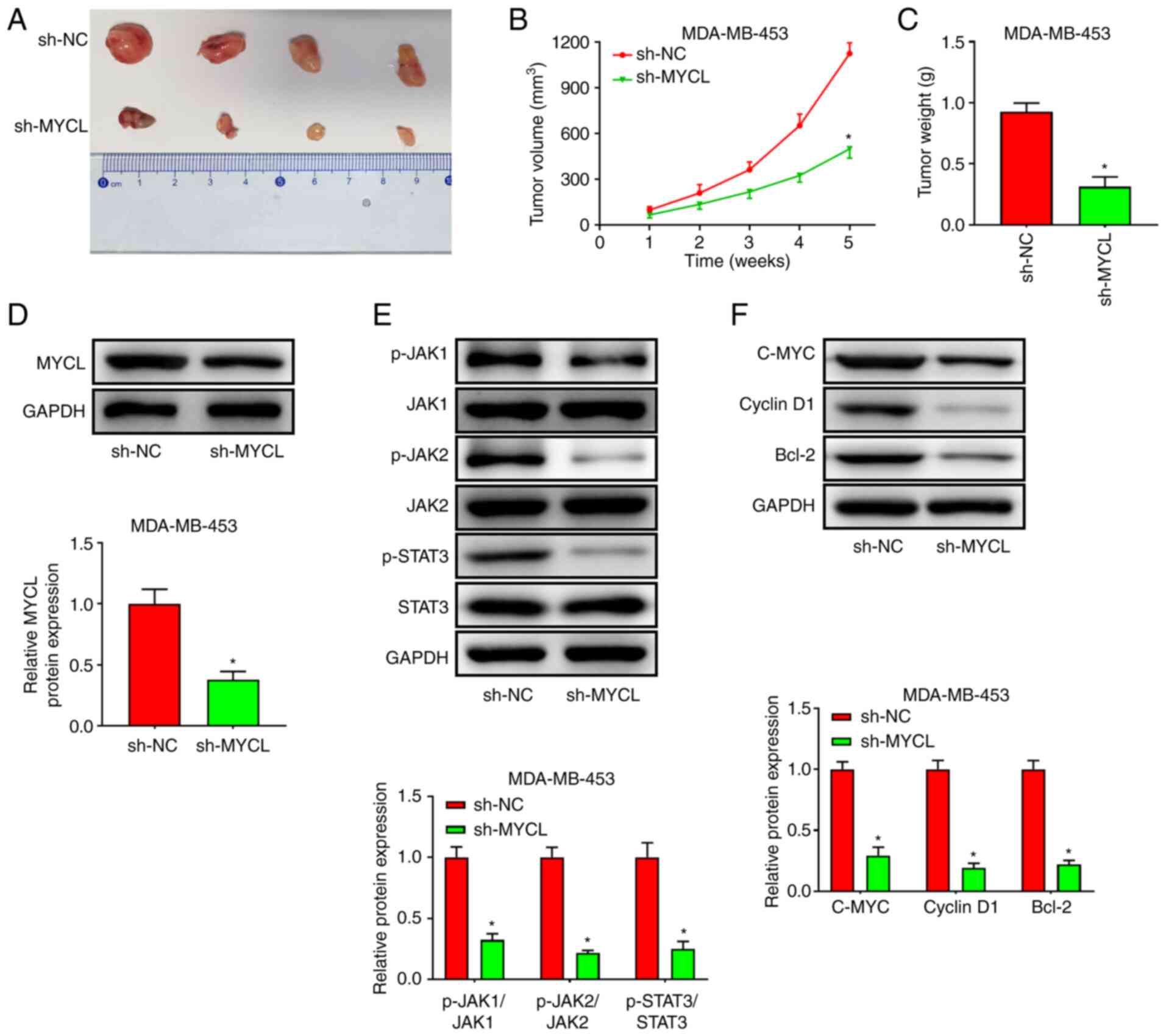
The expression levels of MYCL were higher in
MDA-MB-453 cells than those in MDA-MB-231 cells; therefore,
MDA-MB-453 cells were transfected with sh-MYCL to perform the
xenograft tumor assay. The results revealed that tumor growth was
markedly slower in mice injected with sh-MYCL cells compared with
that in mice injected with sh-NC cells (Fig. 6B). In addition, the weight and size
of the transplanted tumors formed by MDA-MB-453 cells transfected
with sh-MYCL was smaller than that in the sh-NC group (Fig. 6A and C). Notably, western blotting
indicated that MYCL expression was significantly reduced in
xenograft tumors from the sh-MYCL group compared with that in the
tumor tissues of the sh-NC group (Fig.
6D). Additionally, sh-MYCL significantly inhibited the
JAK/STAT3 signaling pathway, as indicated by the significant
decrease in the protein expression levels of p-JAK1/JAK1,
p-JAK2/JAK2, p-STAT3/STAT3, C-MYC, Cyclin D1 and Bcl-2 in the tumor
tissues of the sh-MYCL group compared with those in the sh-NC group
(Fig. 6E and F). Briefly, MYCL
silencing suppressed the growth of TNBC xenograft tumors.
Discussion
TNBC is one of the primary subsets of BC and is
characterized by a lack of ER, PR and HER2 expression (28). TNBC is a type of high-grade
invasive ductal carcinoma with highly aggressive biological
behavior (29). In addition, TNBC
is at a high risk of early recurrence, and is associated with bone,
visceral and brain metastases, low DFS and OS rates, and poor
prognosis (6,30). The evolution of TNBC is a multiple
gene, step and stage pathological process, and an imbalance between
cell growth and death caused by abnormal signal transduction
pathways is an important mechanism underlying the occurrence and
development of TNBC (31).
Therefore, a better understanding of the molecular events
associated with the malignancy may help to understand the
pathological process of TNBC. The present study provided evidence
to support the effect of MYCL expression and the JAK/STAT3 pathway
on the biological behaviors of TNBC, including proliferation,
apoptosis, migration and invasion.
A number of molecular markers have been demonstrated
to regulate different types of cellular processes in TNBC (32). The MYC gene is a common
intracellular proto-oncogene, which is highly expressed in a
variety of tumors, including BC (16). Kato et al (15) reported that MYCL was highly
expressed in SCLC cell lines. Furthermore, inhibition of MYC has
been reported to decrease the proliferation of colon cancer cells
(33). Notably, high expression of
C-MYC protein is associated with a high risk to patients with BC,
and C-MYC protein is considered a tumor marker of TNBC (34). In the present study, TCGA database
analysis demonstrated that MYCL was significantly higher in BC
tumor tissues, particularly in TNBC tumor tissues. The Kaplan-Meier
Plotter analysis also revealed that higher expression of MYCL was
associated with a higher risk of death. Furthermore, the present
results showed that MYCL was markedly upregulated in all TNBC cell
lines compared with in MCF-10A cells.
In recent years, increasing evidence has shown that
MYC genes are extensively distributed in the human genome, and are
involved in cell growth, apoptosis, proliferation and other cell
activities (12,13). In TNBC progression, it has been
reported that epithelial-to-mesenchymal transition (EMT) is
responsible for the migration and invasion of tumor cells via the
acquisition of invasive and mobile capabilities to facilitate
metastasis, which is moderated by numerous related proteins,
including E-cadherin, Vimentin and N-cadherin (35). Cadherins are transmembrane
glycoproteins responsible for cell-cell adhesion and maintenance of
normal tissue architecture (36).
The role of cadherins in the process of cancer development has been
studied widely and numerous studies have described E-cadherin as a
tumor suppressor (37,38). In cancer, a number of tumors have
been shown to exhibit N-cadherin upregulation at the onset of
metastasis (39). Vimentin, a
classic EMT biomarker, is upregulated during EMT in epithelial
cells and induces a mesenchymal phenotype and motor behavior
(40). In the present study,
evidence indicated that silencing MYCL significantly inhibited the
tumorigenesis, invasion and migration of TNBC cells. MYCL silencing
significantly increased E-cadherin expression, and decreased the
expression levels of N-cadherin and Vimentin in TNBC cells.
Conversely, MYCL overexpression enhanced the migratory and invasive
ability of TNBC cells, reduced E-cadherin expression, and increased
the expression levels of N-cadherin and Vimentin. Furthermore, in
vivo animal experiments reinforced the evidence that MYCL silencing
suppressed the growth of TNBC tumors.
Among the most recently recognized carcinogenic
signaling pathways, the JAK/STAT3 signaling pathway has an
important role in the tumorigenesis of BC and other cancer types
(23,41,42).
It has been reported that persistent activation of the JAK/STAT3
pathway could promote tumor malignancy by facilitating tumor cell
immune escape, proliferation, angiogenesis and survival (22,43).
Activation of the JAK/STAT signaling pathway may also lead to
accelerated proliferation of esophageal squamous cell carcinoma
cells (44). Inhibition of the
JAK/STAT3 pathway has been shown to suppress the in vivo growth of
ovarian cancer cells (45). In the
present study, GEO analysis revealed that MYCL activated the
JAK/STAT3 pathway in TNBC. MYCL silencing suppressed the expression
levels of p-JAK1/JAK1, p-JAK2/JAK2 and p-STAT3/STAT3 in TNBC cells.
The downstream proteins of the JAK/STAT3 pathway, C-MYC, Cyclin D1
and Bcl-2, were also suppressed by MYCL silencing. The opposite
results were obtained in MYCL-overexpressing cells. To further
elucidate the association between MYCL and the JAK/STAT3 pathway in
TNBC development, Ruxo, an effective inhibitor of the JAK/STAT3
pathway, was used to treat the transfected MDA-MB-231 cells.
Inhibition of the JAK/STAT3 signaling pathway significantly
suppressed the proliferation and migration of TNBC cells, and
partly abolished the effects of MYCL overexpression on TNBC.
In conclusion, MYCL could promote TNBC progression
by activating the JAK/STAT3 pathway. Therefore, the present study
indicated that MYCL could be regarded as a novel therapeutic target
for the treatment of TNBC. However, there are some limitations in
the present study. Firstly, in future studies, the sample of
patients with TNBC should be collected to support the present
findings; and secondly, the role of MYCL in TNBC and other subtypes
of BC should be further investigated in future studies.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Conception and design: HJ; Perform research: HJ and
XL; Data analysis and interpretation: WW, YH and DR; Manuscript
writing: All authors; Final approval of manuscript: All authors.
All authors have read and approved the final manuscript, and met
the authorship requirements stated earlier in this document, and
each author believes that the manuscript represents honest work. HJ
and DR confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The experimental protocol of the present study was
performed in accordance with the Guide for the Care and Use of
Laboratory Animals and was approved by the Second Hospital of
Shanxi Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BC
|
breast cancer
|
|
ER
|
estrogen receptor
|
|
PR
|
progesterone receptor
|
|
HER2
|
human epidermal growth factor receptor
2
|
|
TNBC
|
triple-negative BC
|
References
|
1
|
Harbeck N, Penault-Llorca F, Cortes J,
Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J and Cardoso F:
Breast cancer. Nat Rev Dis Primers. 5:662019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kyeong S, Cha YJ, Ahn SG, Suh SH, Son EJ
and Ahn SJ: Subtypes of breast cancer show different spatial
distributions of brain metastases. PLoS One. 12:e01885422017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo L, Xie G, Wang R, Yang L, Sun L, Xu M,
Yang W and Chung MC: Local treatment for triple-negative breast
cancer patients undergoing chemotherapy: Breast-conserving surgery
or total mastectomy? BMC Cancer. 21:7172021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Wang SS, Huang H, Cai L, Zhao L,
Peng RJ, Lin Y, Tang J, Zeng J, Zhang LH, et al: Effect of
capecitabine maintenance therapy using lower dosage and higher
frequency vs observation on disease-free survival among patients
with early-stage triple-negative breast cancer who had received
standard treatment: The SYSUCC-001 randomized clinical trial. JAMA.
325:50–58. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burchett JB, Knudsen-Clark AM and Altman
BJ: MYC ran up the clock: The complex interplay between MYC and the
molecular circadian clock in cancer. Int J Mol Sci. 22:77612021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fong KM, Kida Y, Zimmerman PV and Smith
PJ: MYCL genotypes and loss of heterozygosity in non-small-cell
lung cancer. Br J Cancer. 74:1975–1978. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niu F, Dzikiewicz-Krawczyk A, Koerts J, de
Jong D, Wijenberg L, Fernandez Hernandez M, Slezak-Prochazka I,
Winkle M, Kooistra W, van der Sluis T, et al: MiR-378a-3p is
critical for burkitt lymphoma cell growth. Cancers (Basel).
12:35462020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren L, Zhou T, Wang Y, Wu Y, Xu H, Liu J,
Dong X, Yi F, Guo Q, Wang Z, et al: RNF8 induces β-catenin-mediated
c-Myc expression and promotes colon cancer proliferation. Int J
Biol Sci. 16:2051–2062. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lao-On U, Rojvirat P, Chansongkrow P,
Phannasil P, Siritutsoontorn S, Charoensawan V and Jitrapakdee S:
c-Myc directly targets an over-expression of pyruvate carboxylase
in highly invasive breast cancer. Biochim Biophys Acta Mol Basis
Dis. 1866:1656562020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Duffy MJ, O'Grady S, Tang M and Crown J:
MYC as a target for cancer treatment. Cancer Treat Rev.
94:1021542021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang C, Zhang J, Yin J, Gan Y, Xu S, Gu Y
and Huang W: Alternative approaches to target Myc for cancer
treatment. Signal Transduct Target Ther. 6:1172021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ciampricotti M, Karakousi T, Richards AL,
Quintanal-Villalonga À, Karatza A, Caeser R, Costa EA, Allaj V,
Manoj P, Spainhower KB, et al: Rlf-Mycl gene fusion drives
tumorigenesis and metastasis in a mouse model of small cell lung
cancer. Cancer Discov. 11:3214–3229. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kato F, Fiorentino FP, Alibés A, Perucho
M, Sánchez-Céspedes M, Kohno T and Yokota J: MYCL is a target of a
BET bromodomain inhibitor, JQ1, on growth suppression efficacy in
small cell lung cancer cells. Oncotarget. 7:77378–77388. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Zhu C, Tang L, Chen Q, Guan N, Xu K
and Guan X: MYC dysfunction modulates stemness and tumorigenesis in
breast cancer. Int J Biol Sci. 17:178–187. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berger A, Brady NJ, Bareja R, Robinson B,
Conteduca V, Augello MA, Puca L, Ahmed A, Dardenne E, Lu X, et al:
N-Myc-mediated epigenetic reprogramming drives lineage plasticity
in advanced prostate cancer. J Clin Invest. 129:3924–3940. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tjaden B, Baum K, Marquardt V, Simon M,
Trajkovic-Arsic M, Kouril T, Siebers B, Lisec J, Siveke JT, Schulte
JH, et al: N-Myc-induced metabolic rewiring creates novel
therapeutic vulnerabilities in neuroblastoma. Sci Rep. 10:71572020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hassan MS, Cwidak N, Johnson C, Däster S,
Eppenberger-Castori S, Awasthi N, Li J, Schwarz MA and von Holzen
U: Therapeutic potential of the cyclin-dependent kinase inhibitor
flavopiridol on c-Myc overexpressing esophageal cancer. Front
Pharmacol. 12:7463852021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elbadawy M, Usui T, Yamawaki H and Sasaki
K: Emerging roles of C-Myc in cancer stem cell-related signaling
and resistance to cancer chemotherapy: A potential therapeutic
target against colorectal cancer. Int J Mol Sci. 20:23402019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harrison DA: The Jak/STAT pathway. Cold
Spring Harb Perspect Biol. 4:a0112052012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Shea JJ, Schwartz DM, Villarino AV,
Gadina M, McInnes IB and Laurence A: The JAK-STAT pathway: Impact
on human disease and therapeutic intervention. Annu Rev Med.
66:311–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang T, Fahrmann JF, Lee H, Li YJ,
Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al:
JAK/STAT3-regulated fatty acid β-oxidation is critical for breast
cancer stem cell self-renewal and chemoresistance. Cell Metab.
27:136–150.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cascione L, Gasparini P, Lovat F, Carasi
S, Pulvirenti A, Ferro A, Alder H, He G, Vecchione A, Croce CM, et
al: Integrated microRNA and mRNA signatures associated with
survival in triple negative breast cancer. PLoS One. 8:e559102013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the care and use of laboratory animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
28
|
Chen F, Wang Q, Yu X, Yang N, Wang Y, Zeng
Y, Zheng Z, Zhou F and Zhou Y: MCPIP1-mediated NFIC alternative
splicing inhibits proliferation of triple-negative breast cancer
via cyclin D1-Rb-E2F1 axis. Cell Death Dis. 12:3702021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumar P and Aggarwal R: An overview of
triple-negative breast cancer. Arch Gynecol Obstet. 293:247–269.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu Q, Siddharth S and Sharma D: Triple
negative breast cancer: A mountain yet to be scaled despite the
triumphs. Cancers (Basel). 13:36972021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dameri M, Ferrando L, Cirmena G, Vernieri
C, Pruneri G, Ballestrero A and Zoppoli G: Multi-gene testing
overview with a clinical perspective in metastatic triple-negative
breast cancer. Int J Mol Sci. 22:71542021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei LM, Li XY, Wang ZM, Wang YK, Yao G,
Fan JH and Wang XS: Identification of hub genes in triple-negative
breast cancer by integrated bioinformatics analysis. Gland Surg.
10:799–806. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Z, Li K, Zheng Z and Liu Y:
Cordycepin inhibits colon cancer proliferation by suppressing MYC
expression. BMC Pharmacol Toxicol. 23:122022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Constantinou C, Papadopoulos S, Karyda E,
Alexopoulos A, Agnanti N, Batistatou A and Harisis H: Expression
and clinical significance of claudin-7, PDL-1, PTEN, c-Kit, c-Met,
c-Myc, ALK, CK5/6, CK17, p53, EGFR, Ki67, p63 in triple-negative
breast cancer-a single centre prospective observational study. In
Vivo. 32:303–311. 2018.PubMed/NCBI
|
|
35
|
Xu X, Zhang L, He X, Zhang P, Sun C, Xu X,
Lu Y and Li F: TGF-β plays a vital role in triple-negative breast
cancer (TNBC) drug-resistance through regulating stemness, EMT and
apoptosis. Biochem Biophys Res Commun. 502:160–165. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaszak I, Witkowska-Piłaszewicz O,
Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F and Jurka P: Role
of cadherins in cancer-a review. Int J Mol Sci. 21:76242020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bruner H and Derksen PWB: Loss of
E-cadherin-dependent cell-cell adhesion and the development and
progression of cancer. Cold Spring Harb Perspect Biol.
10:a0293302018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kourtidis A, Lu R, Pence LJ and
Anastasiadis PZ: A central role for cadherin signaling in cancer.
Exp Cell Res. 358:78–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cousin H: Cadherins function during the
collective cell migration of xenopus cranial neural crest cells:
Revisiting the role of E-cadherin. Mech Dev. 148:79–88. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu Y, Zhang Y, Sui Z, Zhang Y, Liu M and
Tang H: USP14 de-ubiquitinates vimentin and miR-320a modulates
USP14 and vimentin to contribute to malignancy in gastric cancer
cells. Oncotarget. 8:48725–48736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song X, Liu Z and Yu Z: EGFR promotes the
development of triple negative breast cancer through JAK/STAT3
signaling. Cancer Manag Res. 12:703–717. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu L, Nam S, Tian Y, Yang F, Wu J, Wang
Y, Scuto A, Polychronopoulos P, Magiatis P, Skaltsounis L and Jove
R: 6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and
induces apoptosis of human melanoma cells. Cancer Res.
71:3972–3979. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lv J, Yu W, Zhang Y, Cao X, Han L, Hu H
and Wang C: LNK promotes the growth and metastasis of triple
negative breast cancer via activating JAK/STAT3 and ERK1/2 pathway.
Cancer Cell Int. 20:1242020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fang Y, Zhang S, Yin J, Shen YX, Wang H,
Chen XS and Tang H: LINC01535 promotes proliferation and inhibits
apoptosis in esophageal squamous cell cancer by activating the
JAK/STAT3 pathway. Eur Rev Med Pharmacol Sci. 24:3694–3700.
2020.PubMed/NCBI
|
|
45
|
Wang H and Fu Y: NR1D1 suppressed the
growth of ovarian cancer by abrogating the JAK/STAT3 signaling
pathway. BMC Cancer. 21:8712021. View Article : Google Scholar : PubMed/NCBI
|