Introduction
Nitric oxide (NO) is a significant inducer of
chondrocyte apoptosis, a feature of cartilage degeneration in
diseases such as osteoarthritis (OA) (1–3).
NO-stimulated chondrocytes increased the expression of tumor
suppressor p53 via phosphorylation of the p38 mitogen-activated
protein kinase, resulting in the enhanced transcription of Bax, a
proapoptotic member of the B-cell lymphoma 2 (Bcl-2) family
(4,5). Accumulation of Bax in the mitochondria
leads to cell apoptosis through the release of cytochrome c
from the mitochondria (4). Studies
showing that the reduction of the p53 expression inhibits
NO-induced apoptosis of chondrocytes provide additional evidence of
p53 participation in such apoptosis (4,5).
Endoplasmic reticulum (ER) stress, provoked by an
imbalance between the load of unfolded proteins in the ER and the
capacity of the ER, leads to the accumulation of unfolded or
misfolded proteins in the ER (6).
Mammalian cells induce specialized responses to recover or maintain
ER function by attenuating general translation, upregulating the
expression of ER chaperone proteins, such as the 78 kDa
glucose-regulated protein (GRP78), and by activating the
endoplasmic reticulum-associated protein degradation system
(6). However, if these protective
responses fail and ER stress (ERS) persists, specialized apoptotic
pathways, such as the enhanced expression of C/EBP-homologous
protein (CHOP) and the activation of ER-associated caspase 12, are
activated to eliminate the damaged cells (7). Previous studies showed that articular
chondrocytes in OA cartilage experience ERS during cartilage
degeneration (8,9). ERS induced by pharmacological ERS
inducers such as tunicamycin and thapsigargin may induce apoptosis
of chondrocytes (10). Previously,
we demonstrated that the number of chondrocytes exhibiting ERS
correlates with the number of apoptotic chondrocytes in
osteoarthritic cartilage (11).
Thus, ERS is important in chondrocyte apoptosis during the process
of cartilage degeneration.
In their study, Oliver et al (12) showed that NO is a potent inducer of
ERS in chondrocytes. However, NO has not been demonstrated to
induce sufficient ERS to induce apoptosis in chondrocytes. In
addition, little is known about the correlation between the ERS-
and p53-mediated apoptotic pathways induced by NO in
chondrocytes.
The objective of this study was to determine whether
NO-induced ERS leads to apoptosis in chondrocytes by examining
whether NO-induced apoptosis in cultured chondrocytes was
suppressed by attenuating ERS using the chemical chaperone ERS
inhibitor sodium 4-phenylbutyrate (PBA) or by blocking the
ERS-associated apoptotic pathway with siRNA against CHOP. In
addition, we investigated the temporal relationship between the
expression of CHOP and p53 during chondrocyte apoptosis.
Materials and methods
Reagents
Sodium nitroprusside (SNP), a NO-donor, was
purchased from Sigma (St. Louis, MO, USA). PBA was purchased from
Calbiochem (San Diego, CA, USA). Dulbecco’s modified Eagle’s medium
(DMEM) was purchased from Nacalai Tesuque (Kyoto, Japan). Fetal
bovine serum (FBS), collagenase type II, and
trypsin-ethylenediaminetetraacetic acid (EDTA) were purchased from
Invitrogen (Carlsbad, CA, USA).
Chondrocyte isolation and culture
The animal experiments in this study were designed
according to the Guidelines for Animal Experimentation of Kumamoto
University and were approved by the Animal Experiment Committee of
Kumamoto University. Rat articular chondrocytes were isolated from
slices of femoral head cartilage from 5-week-old Wistar rats (Japan
SLC Inc., Hamamatsu, Japan) by a sequential enzyme digestion method
using collagenase type II as described in a previous study
(13). Isolated chondrocytes were
plated in flasks at a density of 5×104
cells/cm2 in DMEM supplemented with 10% FBS, 100 U/ml
streptomycin and 100 U/ml penicillin. The culture medium was
replaced every 2 days. After 5 days in culture, the cells were
detached using trypsin-EDTA and plated on culture plates at a
density of 10×104 cells/cm2. Three days after
passage, the cells were incubated in serum-free medium with or
without PBA (3 mM) for 12 h and then treated with SNP (0, 0.5, 1 or
2 mM) for 24 h.
siRNA transfection
Stealth Select RNAi specific for Chop
(RSS355093) and Stealth RNAi negative control Hi GC (12935-400)
were purchased from Invitrogen. Rat chondrocytes were seeded at
5×104 cells/cm2 in antibiotic-free medium.
After 24 h, the cells were transfected with 10 nM siRNA duplexes
using lipofectamine RNAiMAX for 36 h (Invitrogen) according to the
manufacturer’s instructions. Following transfection, the cells were
incubated in serum-free medium for 12 h and then treated with SNP
(1 mM) for 24 h.
Nitrite/nitrate assay
Total NO was measured as its breakdown products,
nitrite and nitrate, by the Griess reaction using a nitrate/nitrite
colorimetric assay kit (Cayman, Ann Arbor, MI, USA) according to
the manufacturer’s instructions. Briefly, culture medium from
chondrocytes was reacted with nitrate reductase and its cofactor
for 1 h at room temperature. After color development by the
addition of Griess reagent, the amount of nitrate/nitrite was
determined by absorbance at 540 nm.
Enzyme-linked immunosorbent assay (ELISA)
for apoptosis
The extent of cultured apoptosis of chondrocytes was
analyzed using Cell Death Detection ELISAPLUS (Roche
Applied Science, Mannheim, Germany), according to the
manufacturer’s instructions. Following treatment, the cell lysate
was incubated for 2 h at room temperature with anti-histone-biotin
and anti-DNA-peroxidase antibodies. The absorbance of the samples
was measured at 405 and 490 nm. For each experiment, the amount of
protein in the cell lysate was assessed in separate wells, using
the Quick Start Bradford protein assay (Bio-Rad Laboratories,
Richmond, CA, USA) to normalize the extent of cell apoptosis
(11). For each experiment, the
apoptosis enrichment factor was calculated as the absorbance
(A405–A490 nm) of the cells treated with agents/absorbance
(A405–A490 nm) of untreated control cells.
RNA extraction and real-time polymerase
chain reaction (PCR)
Total RNA was extracted from cultured cells using
the RNeasy mini kit (Qiagen, Valencia, CA, USA) in combination with
DNA digestion using DNase (Qiagen) and reverse-transcribed using
the High Capacity RNA-tocDNA kit (Applied Biosystems, Foster, CA,
USA). The procedures were performed according to the manufacturer’s
instructions. Quantitative real-time RT-PCR analysis was performed
on an Applied Biosystems 7300/7500 Real-time PCR system using the
TaqMan Gene Expression Master mix (Applied Biosystems) and TaqMan
Gene Expression assays for Chop (Rn00492098-g1),
Grp78 (Rn01435771_g1), p53 (Rn00755717_m1), and
Gapdh (Rn99999916_s1) (Applied Biosystems). Reactions were
carried out under the conditions: 2 min at 50°C and 10 min at 95°C;
40 cycles of 15 sec at 95°C and 1 min at 60°C. The relative
quantification of the target gene to Gapdh was calculated
using the ΔΔCt method (User Bulletin no. 2, Applied
Biosystems).
Statistical analysis
Data were expressed relative to the mean value of
cells treated without agents in each experiment. Statistical
analysis was carried out using a one-way analysis of variance
(ANOVA) with the Scheffe’s post hoc tests. P<0.05 was considered
to indicate a statistically significant difference.
Results
SNP induces apoptosis and ERS in
chondrocytes
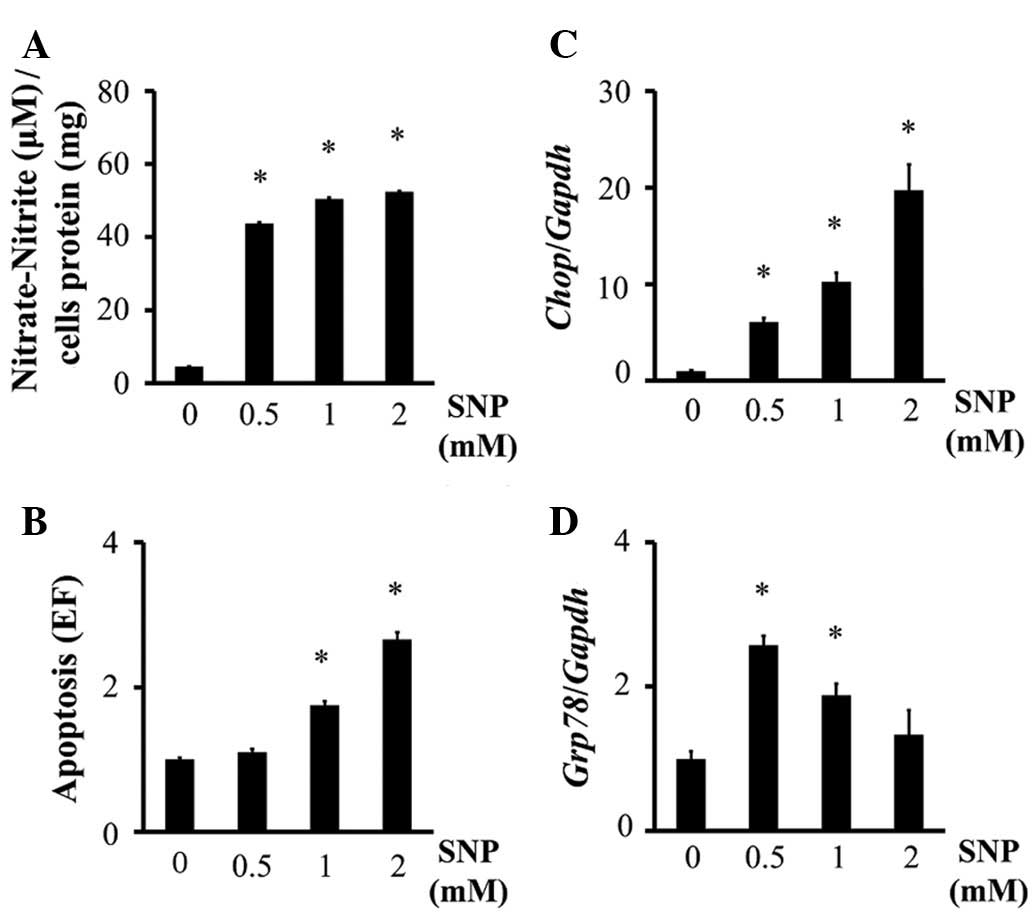
The generation of NO in SNP-treated chondrocytes was
confirmed using the Griess reaction (Fig. 1A). Treating chondrocytes with SNP
significantly increased apoptosis of chondrocytes at doses of ≥1 mM
(Fig. 1B). Chop mRNA
expression increased in SNP-treated chondrocytes in a
dose-dependent manner (Fig. 1C).
SNP also increased Grp78 mRNA expression in chondrocytes
(Fig. 1D), although this effect was
reduced in a dose-dependent manner, and no significant difference
was observed in the level of Grp78 expression between 2 mM
SNP-treated chondrocytes and the control cells (Fig. 1D). These results suggest that NO
induces ERS in chondrocytes. On the basis of these results, 1 mM
SNP was used in subsequent experiments.
ERS inhibitor PBA and Chop knockdown
reduce SNP-induced apoptosis of chondrocytes
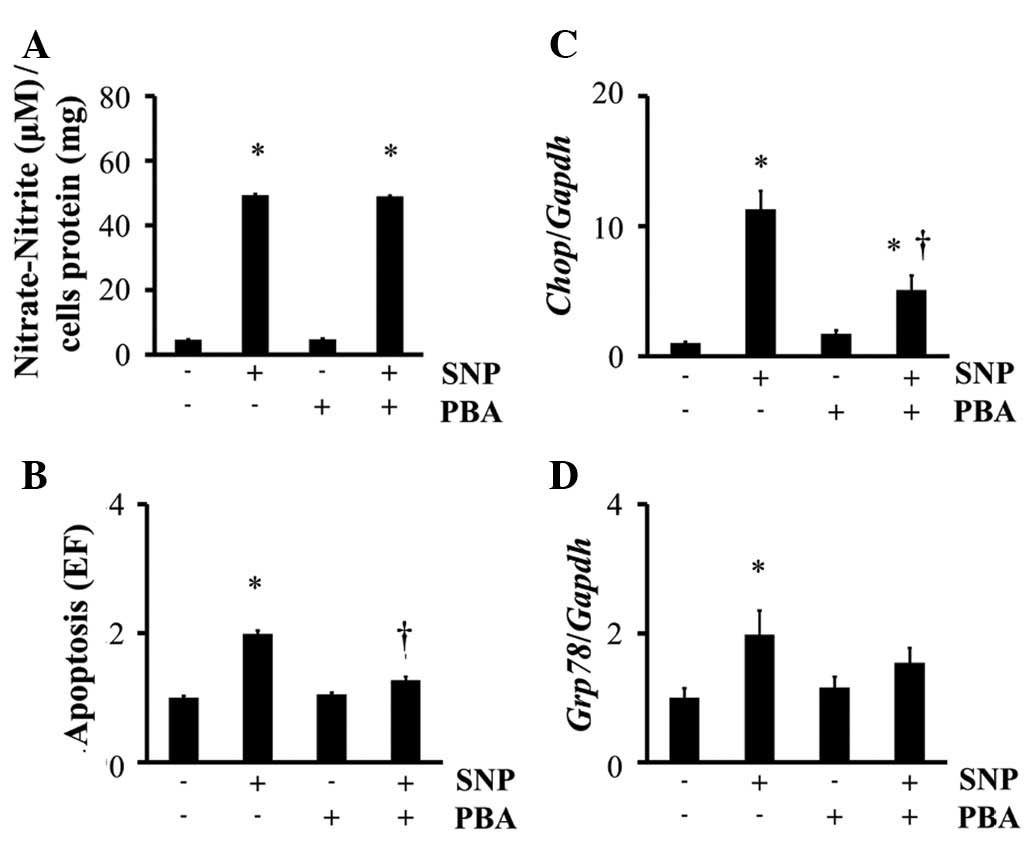
Although PBA had no significant effect on the
generation of NO by SNP (Fig. 2A),
PBA reduced apoptosis (Fig. 2B) and
the mRNA expression of Chop (Fig. 2C) and Grp78 (Fig. 2D) in SNP-treated chondrocytes.
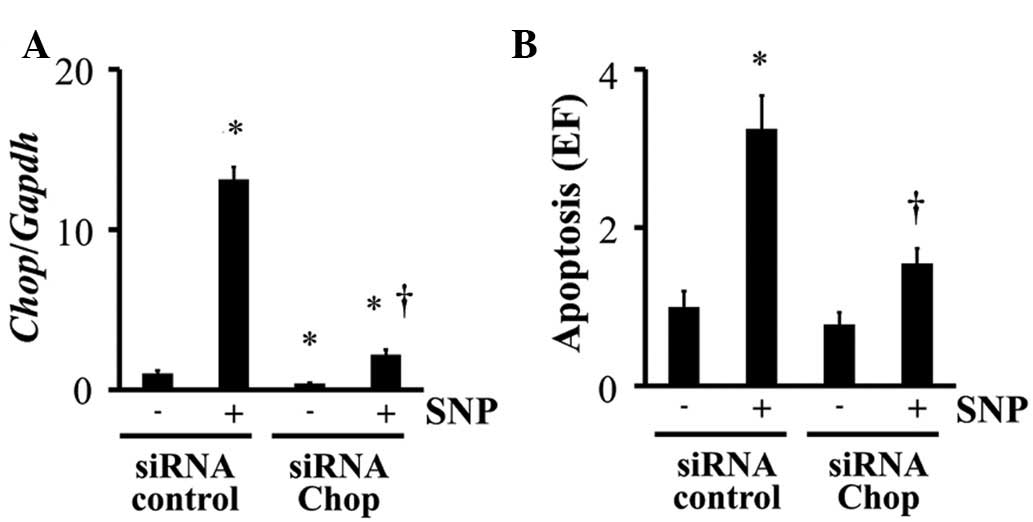
Chop knockdown was performed by siRNA
transfection. Chop siRNA significantly inhibited Chop
expression in chondrocytes, even when the cells were stimulated
with SNP (Fig. 3A). siRNA treatment
also reduced SNP-induced apoptosis of chondrocytes (Fig. 3B).
Temporal relationship between the
expression of Chop and p53 and the induction of apoptosis
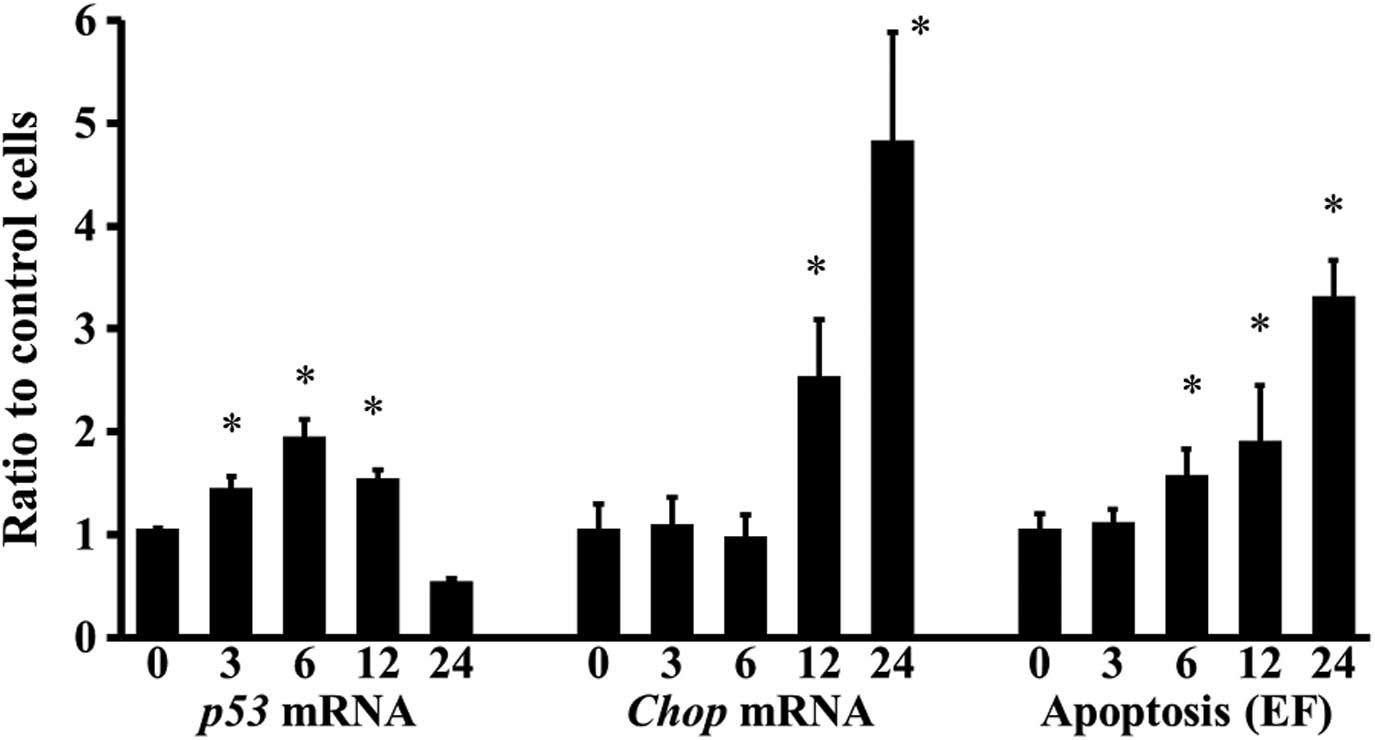
Three hours after chondrocytes were treated with
SNP, p53 expression significantly increased, but Chop
expression and apoptosis were not enhanced (Fig. 4). p53 expression of
chondrocytes achieved a peak 6 h following treatment and was kept
upregulated up to 12 h (Fig. 4).
Chop expression increased in a time-dependent manner 12 h
after SNP stimulation, whereas apoptosis of chondrocytes increased
in a time-dependent manner 6 h after the stimulation (Fig. 4).
Discussion
In this study, we observed that SNP induced
apoptosis of chondrocytes and SNP-stimulated chondrocytes showed an
increase in the CHOP and GRP78 mRNA expression. These results are
consistent with those of previous reports using
S-nitroso-N-acetylpenicillamine (SNAP), another NO
donor (12). Therefore, NO is an
inducer of ERS in chondrocytes. In addition, we found that PBA
treatment as well as the blockade of the CHOP expression was able
to suppress NO-induced apoptosis of chondrocytes.
PBA has been reported to be a chemical chaperone
that reduces the misfolding and mislocalization of mutant
α1-antitrypsin (14) and the
aggregation of Pael-R (15), and
attenuates ERS in liver cells, mouse embryonic fibroblasts
(16) and neuronal cells (15,17).
In a preliminary experiment (data not shown), PBA was confirmed to
also be useful as an ERS inhibitor in chondrocytes, based on the
observation that PBA inhibited the expression of GRP78 and CHOP, as
well as the tunicamycin-stimulated apoptosis in chondrocytes, a
typical ERS inducer. Therefore, the suppressive effect of PBA on
NO-stimulated apoptosis in chondrocytes determined that NO-induced
ERS led chondrocytes to apoptosis. The results of the CHOP
knockdown experiment also supported this conclusion.
In their study, Kim et al (4,5)
demonstrated that p53 was responsible for NO-induced apoptosis of
chondrocytes. In the present study, the p53 expression in
NO-treated chondrocytes increased prior to the induction of
apoptosis and decreased with sustained stimulation by NO, despite
the increased apoptosis. By contrast, the upregulation of CHOP was
induced following the induction of NO-stimulated apoptosis in
chondrocytes. These results suggest that p53 plays a role in the
apoptosis of chondrocytes mainly at the acute stage of NO
stimulation, although this was not the case for ERS. However,
similar to apoptosis, the CHOP expression was found to increase
with sustained stimulation. Qu et al (18) demonstrated that ERS prevented p53
stabilization via glycogen synthase kinase-3β and inhibited
p53-mediated apoptosis. Taking these findings into consideration,
we assumed that ERS initially inhibited p53-mediated NO-induced
apoptosis of chondrocytes, but as NO stimulation progressed, the
persistent impairment altered the ERS responses of chondrocytes
from protective to apoptotic. However, the mechanism of this switch
remains unclear. Therefore, additional studies are required to
clarify the role of ERS in NO-induced apoptosis.
The limitation of this study is that our
observations are based on experiments using exogenous NO generated
by SNP, a NO donor, rather than NO synthases-generated endogenous
NO. However, endogenous NO may also induce ERS and lead to
apoptosis in several cell types, such as pancreatic β cells
(19) and macrophages (20). Therefore, endogenous NO-induced
apoptosis of chondrocytes may also be mediated by ERS.
In conclusion, findings of the present study have
demonstrated that ERS contributed to NO-induced apoptosis of
chondrocytes, using pharmacological attenuation of ERS and blockade
of the ERS-associated apoptotic pathway. However, the contribution
of ERS appears to be limited to persistent impairment of NO
stimulation. Previous studies have demonstrated that NO is the
molecule most responsible for apoptosis of chondrocytes during
cartilage degeneration (1–3). Therefore, our results support that ERS
is involved in apoptosis of chondrocytes of degenerated cartilage.
The importance of ERS in NO-induced apoptosis may provide insight
into the pathology of cartilage degeneration. However, additional
studies are required to investigate the role of ERS in cartilage
biology.
References
|
1
|
Blanco FJ, Ochs RL, Schwarz H and Lotz M:
Chondrocyte apoptosis induced by nitric oxide. Am J Pathol.
146:75–85. 1995.PubMed/NCBI
|
|
2
|
Hashimoto S, Takahashi K, Amiel D, Coutts
RD and Lotz M: Chondrocyte apoptosis and nitric oxide production
during experimentally induced osteoarthritis. Arthritis Rheum.
41:1266–1274. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pelletier JP, Jovanovic DV, Lascau-Coman
V, Fernandes JC, Manning PT, Connor JR, Currie MG and
Martel-Pelletier J: Selective inhibition of inducible nitric oxide
synthase reduces progression of experimental osteoarthritis in
vivo: possible link with the reduction in chondrocyte apoptosis and
caspase 3 level. Arthritis Rheum. 43:1290–1299. 2000. View Article : Google Scholar
|
|
4
|
Kim SJ, Hwang SG, Shin DY, Kang SS and
Chun JS: p38 kinase regulates nitric oxide-induced apoptosis of
articular chondrocytes by accumulating p53 via NFkappa B-dependent
transcription and stabilization by serine 15 phosphorylation. J
Biol Chem. 277:33501–33508. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim SJ, Ju JW, Oh CD, Yoon YM, Song WK,
Kim JH, Yoo YJ, Bang OS, Kang SS and Chun JS: ERK-1/2 and p38
kinase oppositely regulate nitric oxide-induced apoptosis of
chondrocytes in association with p53, caspase-3, and
differentiation status. J Biol Chem. 277:1332–1339. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gotoh T and Mori M: Nitric oxide and
endoplasmic reticulum stress. Arterioscler Thromb Vasc Biol.
26:1439–1446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Horton WE Jr, Bennion P and Yang L:
Cellular, molecular, and matrix changes in cartilage during aging
and osteoarthritis. J Musculoskelet Neuronal Interact. 6:379–381.
2006.PubMed/NCBI
|
|
9
|
Nugent AE, Speicher DM, Gradisar I,
McBurney DL, Baraga A, Doane KJ and Horton WE Jr: Advanced
osteoarthritis in humans is associated with altered collagen VI
expression and upregulation of ER-stress markers Grp78 and bag-1. J
Histochem Cytochem. 57:923–931. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang L, Carlson SG, McBurney D and Horton
WE Jr: Multiple signals induce endoplasmic reticulum stress in both
primary and immortalized chondrocytes resulting in loss of
differentiation, impaired cell growth, and apoptosis. J Biol Chem.
280:31156–31165. 2005. View Article : Google Scholar
|
|
11
|
Takada K, Hirose J, Senba K, Yamabe S,
Oike Y, Gotoh T and Mizuta H: Enhanced apoptotic and reduced
protective response in chondrocytes following endoplasmic reticulum
stress in osteoarthritic cartilage. Int J Exp Pathol. 92:232–242.
2011. View Article : Google Scholar
|
|
12
|
Oliver BL, Cronin CG, Zhang-Benoit Y,
Goldring MB and Tanzer ML: Divergent stress responses to IL-1beta,
nitric oxide, and tunicamycin by chondrocytes. J Cell Physiol.
204:45–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hirose J, Ryan LM and Masuda I:
Up-regulated expression of cartilage intermediate-layer protein and
ANK in articular hyaline cartilage from patients with calcium
pyrophosphate dihydrate crystal deposition disease. Arthritis
Rheum. 46:3218–3229. 2002. View Article : Google Scholar
|
|
14
|
Burrows JA, Willis LK and Perlmutter DH:
Chemical chaperones mediate increased secretion of mutant alpha
1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy
for prevention of liver injury and emphysema in alpha 1-AT
deficiency. Proc Natl Acad Sci USA. 97:1796–1801. 2000. View Article : Google Scholar
|
|
15
|
Kubota K, Niinuma Y, Kaneko M, Okuma Y,
Sugai M, Omura T, Uesugi M, Uehara T, Hosoi T and Nomura Y:
Suppressive effects of 4-phenylbutyrate on the aggregation of Pael
receptors and endoplasmic reticulum stress. J Neurochem.
97:1259–1268. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qi X, Hosoi T, Okuma Y, Kaneko M and
Nomura Y: Sodium 4-phenylbutyrate protects against cerebral
ischemic injury. Mol Pharmacol. 66:899–908. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qu L, Huang S, Baltzis D, Rivas-Estilla
AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A and
Koromilas AE: Endoplasmic reticulum stress induces p53 cytoplasmic
localization and prevents p53-dependent apoptosis by a pathway
involving glycogen synthase kinase-3beta. Genes Dev. 18:261–277.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oyadomari S, Takeda K, Takiguchi M, Gotoh
T, Matsumoto M, Wada I, Akira S, Araki E and Mori M: Nitric
oxide-induced apoptosis in pancreatic beta cells is mediated by the
endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA.
98:10845–10850. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gotoh T, Oyadomari S, Mori K and Mori M:
Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated
by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J
Biol Chem. 277:12343–12350. 2002. View Article : Google Scholar : PubMed/NCBI
|