Introduction
Prader-Willi syndrome (PWS) is an imprinting
disorder, which arises due to three main mechanisms, and eventually
results in total absence of the paternally imprinted genes
expression in the 15q11-q13 region. The three genetic mechanisms
are paternal deletion of this region (in <70% of cases),
maternal uniparental disomy (UPD) (in 25–30%) and imprinting center
defect (in 2–5%) (1–3). The paternal copies of the genes are
typically expressed in the PWS region; however, due to
parent-of-origin-specific imprinting, the maternal copies of these
genes are silenced. Almond-shaped and occasional upslanting
palpebral fissures, bitemporal narrowing and strabismus are
frequent characteristic facial features of individuals with PWS
(2,4).
Hypogonadism, which manifests as genital hypoplasia (including
cryptorchidism) and delayed or incomplete pubertal development, is
another characteristic feature (5).
With regards to neurobehavior features, PWS is characterized by
decreased fetal movement, neonatal hypotonia and feeding
difficulties, which lead to the failure to thrive in the postnatal
period. The majority of individuals with PWS have mild intellectual
disability, particularly behavioral problems, including
manipulative behavior, obsessive-compulsive behaviors, compulsive
skin picking, stubbornness and temper tantrums. Attention-deficit
and hyperactivity symptoms may also occur, along with features
suggestive of autism spectrum disorders (2,4,6).
Concerns regarding assisted reproductive
technologies (ART) have been raised due to the possible
associations between the safety and genetic disorders, particularly
imprinting defects (7). Previous
studies have provided evidence for an association between
imprinting disorders and ARTs. Nine imprinting syndromes have been
reported as associated with ART (7,8), whereas
other studies have reported no correlation (9,10). For
example, Angelman syndrome (AS) and Beckwith-Wiedemann syndrome are
two disorders in which an imprinting defect accounts for a
significant proportion of affected individuals, with known
increased risks for patients born following ART (11–13).
The present study reports a case of a 6-year-old
girl with PWS conceived following an ART pregnancy, who presented
with the clinical features of PWS. Molecular analysis confirmed a
de novo microdeletion between breaking point 2 (BP2) and
BP3, which occurs from makorin ring finger protein 3 (MKRN3)
through HECT and RLD domain containing E3 ubiquitin protein ligase
2 (HERC2) at 15q11.2-q13.1.
Case report
Patient and clinical findings
The patient was a 6-year-old female who was the
product of a dizygotic twin pregnancy preceded by in vitro
fertilization (IVF). The parents and twin sister were healthy with
a normal level of intelligence. Family history was negative for
mental retardation, behavioral problems and congenital
abnormalities. All the subjects provided written informed consent
for clinical and molecular analyses, and the study protocol was
approved by the Institutional Review Board (DC15ZISE0114) of The
Catholic University of Korea, Daejeon St. Mary's Hospital (Seoul
Korea). The determination of twin zygosity was identified by a
short tandem repeat (STR) multiplex assay (AmpFLSTR®
Identifiler; Applied Biosystems, Foster City, CA, USA) that
amplifies 15 tetranucleotide repeat loci for autosomal, codominant,
unlinked loci and the gender-determining marker amelogenin in a
single polymerase chain reaction (PCR) amplification. STR analysis
also confirmed the biological association of the father and mother
with the proband. Pregnancy was complicated due to small size for
the gestational age and delivery was at 31 weeks of gestation. The
birth weight of the patient was 1,030 g (below 10th percentile),
length was 38 cm (25th percentile) and head circumference was 28 cm
(25th percentile). Marked lethargy with no crying and poor reflexes
at birth was evident. The patient was intubated immediately and
ventilator care was required for 4 weeks. The patient had neonatal
feeding difficulties necessitating gavage tube feeding to assure
adequate nutrition until 2 months of age. No abnormalities were
evident on echocardiogram, urogeninal sonogram, audiometric test
and brain magnetic resonance imaging. Hypotonia was severe, but
gradually improved with age. Sucking power slowly improved over
several months and normal eating was possible at 12 months of age.
Among laboratory analysis in the first year, biochemical analysis
including blood electrolytes, liver and renal function tests,
complete blood count, thyroid function tests, serological tests for
TORCH infections, amino acid chromatography, transfontanelle
ultrasonography, electroencephalography and electromyography were
within the normal limits. Motor milestones and language development
were delayed; the patient walked independently at 22 months of age
and could speak in sentences at 3.5 years of age. Hyperphagia
occurred at 2 years of age and obesity followed at 3 years of age.
The patient had a central obesity with slender arms and legs and
had abnormal lipid profiles, with mild hypercholesterolemia (total
cholesterol, 219 mg/dl; normal range, 120–180 mg/dl) and
hypertriglycemia (triglycerides, 346 mg/dl; normal range 35–110
mg/dl). Facial appearance was characteristic with narrow bifrontal
diameter, short upturned nose and downturned mouth with a thin lip.
The patient had whitish skin and brown hairs. Upper extremities
were notable for the small hands relative to body size. Short
stature persisted following birth (below 3rd percentile), but
endocrinological investigations including follicle-stimulating
hormone, luteinizing hormone, prolactin, human growth hormone,
adrenocorticotropic hormone, cortisol, free triiodothyronine, free
thyroxine, thyroid-stimulating hormone and insulin were within
normal ranges. The patient was borderline mental retardation with
an IQ of 75 at 6 years of age. The patient did not present with
behavioral problems including temper tantrums, violent behavior and
obsessive/compulsive behavior, anxiety or depression. The patient
had mild learning disabilities, and so attended a kindergarten for
children with normal development. However, the patient received
additional educational programs including speech therapy, regular
exercise and social skill training. Due to the phenotypic
resemblance and the medical records, which were highly indicative
of PWS, methylation-specific multiplex ligation-dependent probe
amplification (MS-MLPA) and array-based comparative genome
hybridization (aCGH) analyses detected a de novo
microdeletion involving BP2 and BP3 (type 2) at 15q11.2-q13.
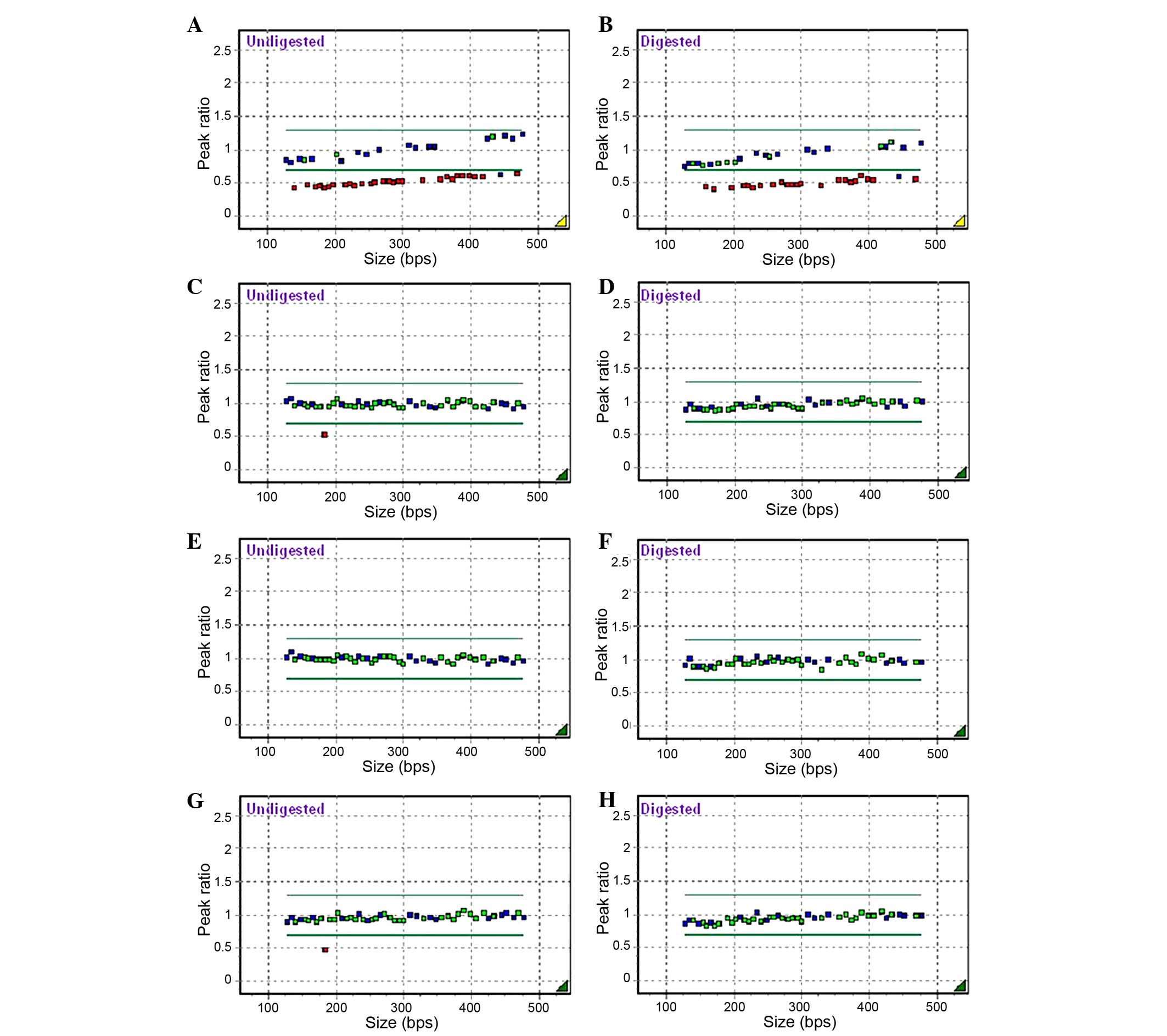
MS-MLPA
MS-MLPA was performed using a MS-MLPA probemix
ME028-B2 Prader-Willi/Angelman kit (MRC-Holland, Amsterdam, The
Netherlands) was performed according to the manufacturer's protocol
in the proband and family members (Fig.
1). A total of 32 probes specific for sequences in or near the
PWS/AS critical region of chromosome 15q11 were used to detect copy
number changes, as well as to analyze CpG island methylation of the
15q11 region for the presence of aberrant methylation patterns
either caused by UPD or by imprinting defects in a
semi-quantitative manner. The manufacturer's protocols were
followed for the DNA preparation, probe hybridization, probe
ligation, enzyme digestion and multiplex PCR reaction. Capillary
electrophoresis and fragment analysis were conducted on an ABI 3130
DNA analyzer (Applied Biosystems). The resulting peak intensities
were normalized to the manufacturer's control probes and to normal
DNA as a reference. A probe-peak ratio between 0.7 and 1.3 was
considered to represent a normal copy number (wild-type), and a
ratio between 0.3 and 0.7 represented a loss of one copy number
(deletion). To determine the methylation status, the normalized
probe-peak ratio of a ligation-treated sample was compared with the
ratio of the same sample treated with ligation and restriction
digestion (by HhaI), using the three ligation control
probes, four methylation-sensitive probes located on the
SNRPN promoter region and one located on the NDN
promoter region. MS-MLPA revealed a de novo microdeletion
from MKRN3 on 15q11.2 to GABRB3 on 15q12 while
sparing APBA2 (there were no MS-MLPA probes for
GABRA5, GABRG3, OCA2, and HERC2, which
are located between GABRB3 on 15q12 and APBA2 on
15q13.1) in the proband only (Fig. 1A and
B).
aCGH
The MS-MLPA result from the proband was verified by
a genomic microarray using the SurePrint G3 Human CGH+SNP
Microarray 4×180K kit (Agilent Technologies, Inc., Santa Clara, CA,
USA) according to the manufacturer's protocol. As a reference
sample, the male or female genomic DNA from Agilent was used. The
microarray slides were scanned at 3-micron resolution on an Agilent
microarray scanner and the raw data were extracted using the
Agilent Feature Ex traction software V10.7.3.1. Raw data were
analyzed using Agilent Genomic Workbench software, CGH module
7.0.4.0 (Agilent Technologies). Copy number alterations (CNAs) were
reported based on the following criteria: Amplifications and
deletions were scored when there was a 10-probe call with a minimum
absolute average log2 ratio of 0.25, minimum genomic
sizes of 0.5 Mb and <50% overlap with known CNAs (Database of
Genomic Variants; http://dgv.tcag.ca/dgv/app/home). Mosaicism,
coexisting minor populations with major diploid population, was
detected by visual inspection according to the following criteria:
i) A discontinuous line in the copy number state window, compared
with a continuous consistent line representing the major clonal
population; ii) intermediate values in the smooth signal, such as a
minimum of 10 markers with a minimum absolute average
log2 ratio of 0.1. Copy neutral-loss of heterozygosity
(LOH) >5 Mb was considered using the LOH algorithm at the
default threshold of 6.0. Consequently, the aCGH demonstrated a
deletion of ~4.8 Mb extending from MKRN3 through
HERC2 at 15q11.2-q13. Therefore, this subject had a type 2
deletion involving BP2 proximally and BP3 distally (Fig. 2).
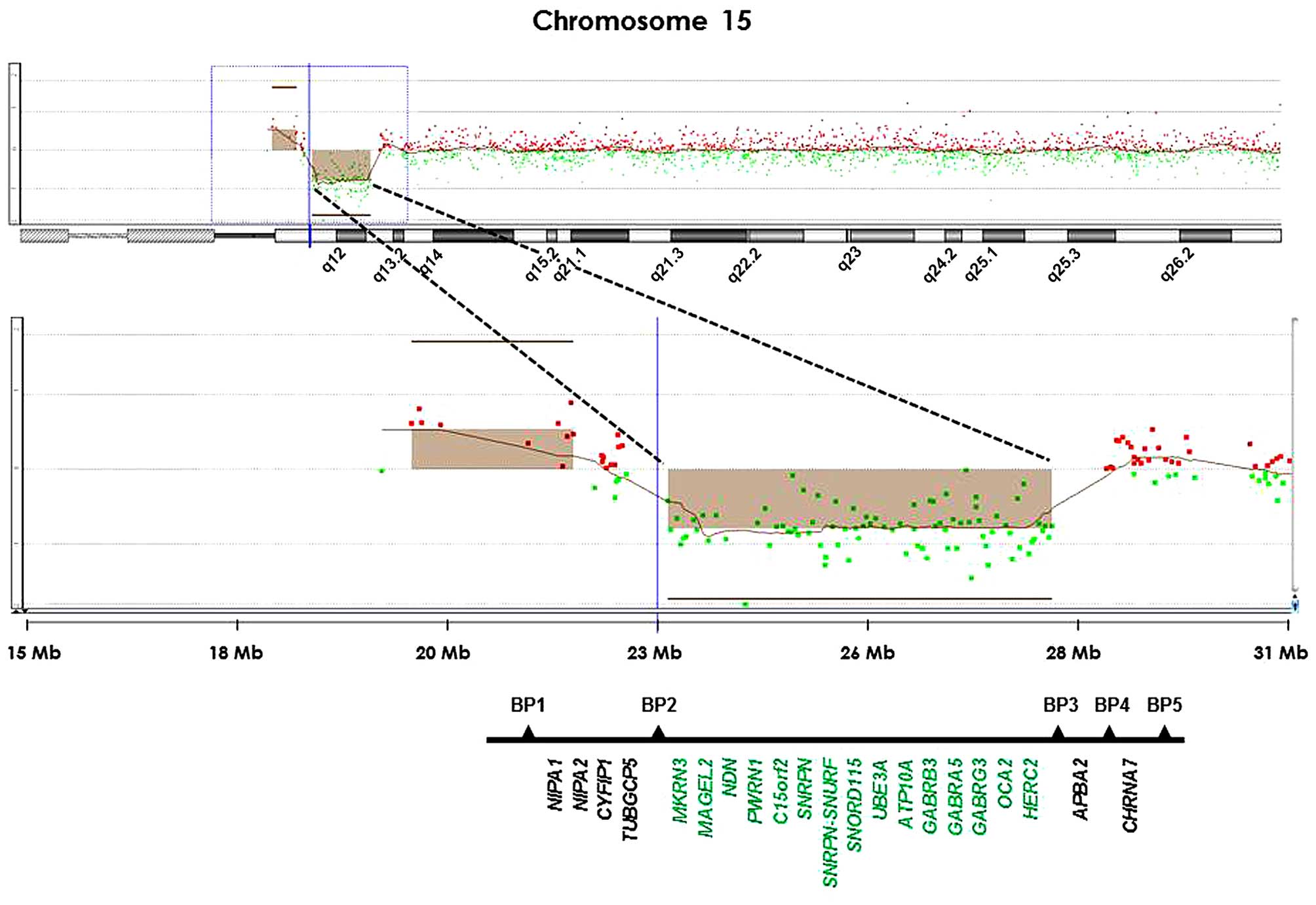 | Figure 2.A high resolution oligonucleotide
array-based comparative genome hybridization plot is shown with
loss of a segment in 15q11.2–15q13.1 from position 23,699,701 to
28,525,460 base pairs (green dots) in the proband. The deleted
segment is with respect to MKRN3, MAGEL2, NDN,
PWRN1, C15orf2, SNRPN,
SNRPN-SNURF, SNORD115, UBE3A,
ATP10A, GABRB3, GABRA5, GABRG3,
OCA2 and HERC2 within the interval (in green). |
Discussion
The present study reports the case of a 6-year-old
female with PWS caused by a de novo microdeletion at
15q11.2-q13.1, which is, to the best of our knowledge, the first
PWS case born following ART reported in South Korea. MS-MLPA and
aCGH demonstrate type 2 microdeletion between BP2 and BP3 occurring
from MKRN3 through HERC2 at 15q11.2-q13.1. MS-MLPA
reliably detected an approximation of the BPs and deletion size as
evidenced by agreement with aCGH in the present case. Compared with
aCGH, the MS-MLPA technique was much more labor and cost-effective,
although aCGH provides more precise information regarding the
extent of the deletion. In addition, the DNA methylation component
of MS-MLPA allows differentiation between PWS and AS, and maternal
and paternal 15q11.2 duplications, as well as between uniparental
and biparental disomy. Therefore, MS-MLPA is recommended as the
first screening test when considering PWS or AS based on clinical
criteria.
The microdeletion class in PWS is typically
subdivided into type 1 (BP1-BP3) and type 2 (BP2-BP3) based on
their proximal breakpoints (14). Type
1 and 2 microdeletions are almost always de novo events.
Type 1 microdeletions have been reported to be associated with
worse adaptive behavior, more severe compulsive behavior and more
impairments in reading, math skills and visual perception than
those with type 2; the present case could support type 2
microdeletion (15). However, several
studies have investigated phenotypic characteristics between type 1
and 2 microdeletions in PWS, and there has been a lack of consensus
among the different studies (15,16).
Furthermore, the subjects with a unique or an atypical
microdeletion revealed distinct phenotypic features (17). Kim et al (17) suggested that the microdeletions in PWS
should be characterized by accurately determining their proximal
and distal BPs, rather than just their proximal BP, as the distal
BPs were not always well delineated in a number of these studies.
By contrast, individuals with PWS due to maternal UPD are less
severely affected. They have higher verbal IQ and milder physical
features than those with microdeletions (2,18).
Whether ART has adverse effects on the fetal genetic
status remains to be elucidated. However, a number of complications
including imprinting defects have been reportedly associated with
ART (19). An increased incidence of
aneuploidy and de novo sex chromosome aberrations have been
reported previously (20,21) and whether or not an increased risk for
imprinting disorders exists remains to be elucidated (22,23). A
possible link between ARTs and genomic imprinting disorders has
been reported, particularly in AS and Beckwith-Wiedemann syndrome
(22). They are mainly caused by four
mechanisms: Large deletions or duplications of chromosomal regions
that contain imprinted genes, UPD, imprinting mutations and
epimutation (24).
Although children with PWS are more likely to be
born to parents with fertility problems (22), no significant associations have been
described between the incidences of the PWS and ART, such as IVF or
intracytoplasmic sperm injection. Several studies found no
association between ART and PWS as paternal deletions and maternal
UPD account for the majority of PWS cases (13,25,26). A previous study also suggested that the
proportion of ART births is not associated with an increased risk
of PWS, but did identify a significantly increased proportion of
maternal UPD and imprinting defects in the ART-conceived PWS study
population (27). As older parents may
have experienced infertility issues due to advanced parental ages,
maternal UPD is associated with increasing maternal age, so the
ART-conceived PWS may be affected due to mechanisms causing UPD and
is not due to the ART procedures themselves (28,29).
In conclusion, genotype-phenotype counseling is
important for estimating PWS severity due to type 1 or 2, as well
as unique, microdeletions due to a novel distal and/or proximal BP.
Molecular analyses, including MS-MLPA as a screening method and
aCGH as a confirm test, would be more beneficial for the diagnosis
and prognosis of PWS. In addition to previous studies, the present
study contributes to the consensus regarding genotype-phenotype
comparisons in this respect. Further studies regarding the safety
of ART are required to elucidate a possible causal association
between 15q microdeletion and ART.
Acknowledgements
The authors are grateful to The Catholic Genetic
Laboratory Center for their assistance in performing the present
study and compiling this report. The study was supported by
Samkwang Medical Laboratories (Seoul, Republic of Korea).
References
|
1
|
Butler MG: Prader-Willi syndrome: Obesity
due to genomic imprinting. Curr Genomics. 12:204–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cassidy SB and Driscoll DJ: Prader-Willi
syndrome. Eur J Hum Genet. 17:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Horsthemke B and Wagstaff J: Mechanisms of
imprinting of the Prader-Willi/Angelman region. Am J Med Genet A.
146A:2041–2052. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Whittington J and Holland T: Prader-Willi
syndrome: Development and manifestations. Cambridge University
Press. Cambridge, UK: 2004. View Article : Google Scholar
|
|
5
|
Gunay-Aygun M, Schwartz S, Heeger S,
O'Riordan MA and Cassidy SB: The changing purpose of Prader-Willi
syndrome clinical diagnostic criteria and proposed revised
criteria. Pediatrics. 108:E922001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Butler MG, Hanchett JM and Thompson T:
Clinical findings and natural history of Prader-Willi syndrome.
Management of Prader-Willi Syndrome (3rd). Butler MG, Lee PDK and
Whitman BY: Springer. (New York, NY). 3–48. 2006. View Article : Google Scholar
|
|
7
|
Laprise SL: Implications of epigenetics
and genomic imprinting in assisted reproductive technologies. Mol
Reprod Dev. 76:1006–1018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Savage T, Peek J, Hofman PL and Cutfield
WS: Childhood outcomes of assisted reproductive technology. Hum
Reprod. 26:2392–2400. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bowdin S, Allen C, Kirby G, Brueton L,
Afnan M, Barratt C, Kirkman-Brown J, Harrison R, Maher ER and
Reardon W: A survey of assisted reproductive technology births and
imprinting disorders. Hum Reprod. 22:3237–3240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lidegaard O, Pinborg A and Andersen AN:
Imprinting diseases and IVF: Danish National IVF cohort study. Hum
Reprod. 20:950–954. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cox GF, Bürger J, Lip V, Mau UA, Sperling
K, Wu BL and Horsthemke B: Intracytoplasmic sperm injection may
increase the risk of imprinting defects. Am J Hum Genet.
71:162–164. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maher ER: Imprinting and assisted
reproductive technology. Hum Mol Genet. 14(Suppl 1): R133–R138.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sutcliffe AG, Peters CJ, Bowdin S, Temple
K, Reardon W, Wilson L, Clayton-Smith J, Brueton LA, Bannister W
and Maher ER: Assisted reproductive therapies and imprinting
disorders - a preliminary British survey. Hum Reprod. 21:1009–1011.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Butler MG, Fischer W, Kibiryeva N and
Bittel DC: Array comparative genomic hybridization (aCGH) analysis
in Prader-Willi syndrome. Am J Med Genet A. 146A:854–860. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Butler MG, Bittel DC, Kibiryeva N,
Talebizadeh Z and Thompson T: Behavioral differences among subjects
with Prader-Willi syndrome and type I or type II deletion and
maternal disomy. Pediatrics. 113:565–573. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dykens EM and Roof E: Behavior in
Prader-Willi syndrome: Relationship to genetic subtypes and age. J
Child Psychol Psychiatry. 49:1001–1008. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SJ, Miller JL, Kuipers PJ, et al:
Unique and atypical deletions in Prader-Willi syndrome reveal
distinct phenotypes. Eur J Hum Genet. 20:283–290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roof E, Stone W, MacLean W, Feurer ID,
Thompson T and Butler MG: Intellectual characteristics of
Prader-Willi syndrome: Comparison of genetic subtypes. J Intellect
Disabil Res. 44:25–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iliadou AN, Janson PC and Cnattingius S:
Epigenetics and assisted reproductive technology. J Intern Med.
270:414–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonduelle M, Van Assche E, Joris H,
Keymolen K, Devroey P, Van Steirteghem A and Liebaers I: Prenatal
testing in ICSI pregnancies: Incidence of chromosomal anomalies in
1586 karyotypes and relation to sperm parameters. Hum Reprod.
17:2600–2614. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kushnir VA and Frattarelli JL: Aneuploidy
in abortuses following IVF and ICSI. J Assist Reprod Genet.
26:93–97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harper J, Geraedts J, Borry P, Cornel MC,
Dondorp WJ, Gianaroli L, Harton G, Milachich T, Kääriäinen H,
Liebaers I, et al: ESHG, ESHRE and EuroGentest2: Current issues in
medically assisted reproduction and genetics in Europe: Research,
clinical practice, ethics, legal issues and policy. Hum Reprod.
29:1603–1609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vermeiden JP and Bernardus RE: Are
imprinting disorders more prevalent after human in vitro
fertilization or intracytoplasmic sperm injection? Fertil Steril.
99:642–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amor DJ and Halliday J: A review of known
imprinting syndromes and their association with assisted
reproduction technologies. Hum Reprod. 23:2826–2834. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Doornbos ME, Maas SM, McDonnell J,
Vermeiden JP and Hennekam RC: Infertility, assisted reproduction
technologies and imprinting disturbances: A Dutch study. Hum
Reprod. 22:2476–2480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Neri QV, Takeuchi T and Palermo GD: An
update of assisted reproductive technologies results in the United
States. Ann N Y Acad Sci. 1127:41–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gold JA, Ruth C, Osann K, Flodman P,
McManus B, Lee HS, Donkervoort S, Khare M, Roof E, Dykens E, et al:
Frequency of Prader-Willi syndrome in births conceived via assisted
reproductive technology. Genet Med. 16:164–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Whittington JE, Butler JV and Holland AJ:
Changing rates of genetic subtypes of Prader-Willi syndrome in the
UK. Eur J Hum Genet. 15:127–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Butler MG, Sturich J, Myers SE, Gold JA,
Kimonis V and Driscoll DJ: Is gestation in Prader-Willi syndrome
affected by the genetic subtype? J Assist Reprod Genet. 26:461–466.
2009. View Article : Google Scholar : PubMed/NCBI
|