Introduction
Cardiac hypertrophy is the response to stress or
disease, such as hypertension, myocardial infarction (MI) and
valvular heart disease. The incidence of cardiac hypertrophy is
~0.2% in adults. It is not gender, age, race or geographic specific
(1). It causes sudden cardiac death in
young patients (2) and develops into
heart failure in the elderly. Heart failure may be responsible for
as many as 60% of cardiac hypertrophy-associated deaths. Patients
with cardiac hypertrophy are reported to have a mortality rate of
~1.0% per year (3). The features of
cardiac hypertrophy include enlarged cardiomyocyte size, increased
protein synthesis, elevated fetal gene atrial natriuretic peptide,
brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC)
expression, and abnormal sarcomeric organization, as well as
heightened expression of fibronectin. There are numerous cell
signaling pathways associated with cardiac hypertrophy and cardiac
failure, including signal transducer and activator of transcription
3, Akt, extracellular signal-regulated kinase (ERK)1/2 and liver
kinase B1/5′ AMP-activated protein kinase (4–6).
Resistin is an adipocyte-secreted adipokine, which
has been linked to obesity, diabetes, insulin resistance and
cardiac hypertrophy (7,8). Loss of resistin has been shown to improve
insulin sensitivity (9). In addition,
treatment with resistin causes glucose intolerance (10). Resistin is regulated by cytokines,
including endothelin, insulin, insulin-like growth factors and
peroxisome proliferator-activated receptor γ (11,12). In
rodents, resistin is located within adipocytes, while human
resistin is primarily expressed in macrophages and neutrophils
(13–15). Human resistin is released in response
to inflammatory stimuli (16), and is
found at elevated levels in autoimmune disease and sepsis (17). Previous studies have shown that
resistin impairs cardiomyocyte glucose handling and induces cardiac
hypertrophy (8,10).
Apelin is an endogenous peptide ligand for the G
protein-coupled apelin receptor (APJ). Apelin and APJ are expressed
in the heart (18,19). Apelin is synthesized as a 77-amino acid
peptide processed into various C-terminal fragments, including
apelin-36, −19, −17, −13, −12, and [Pyr1]-apelin-13. Apelin-13 is
the most stable amongst them (20,21).
Deficiency of apelin exacerbates MI adverse remodeling and
ischemia-reperfusion (I/R) injury (21). Treatment with apelin promotes
myocardial angiogenesis and improves cardiac function in post-MI
mice (22). Recent studies have shown
that apelin ameliorates high fat diet-induced cardiac hypertrophy
(23). Although resistin has been
reported to induce cardiac hypertrophy, while apelin is reported to
inhibit cardiac hypertrophy, it is not known whether apelin
inhibits resistin-induced cardiomyocyte hypertrophy via
inactivation of the ERK signaling pathway.
The aim of the current study was to investigate the
effects of apelin on ERK cell signaling in the inhibition of
resistin-induced cardiomyocyte hypertrophy in H9c2 cells.
Materials and methods
Reagents
Apelin-13 was obtained from Phoenix Pharmaceuticals,
Inc. (Burlingame, CA, USA). Recombinant human resistin was
purchased from Peprotech (Rocky Hill, NJ, USA). The H9c2 cells (rat
cardiomyoblast cells) were obtained from the American Type Culture
Collection (Manassas, VA, USA). Fetal calf serum was purchased from
Zhejiang Tianhang Biological Technology (Huzhou, Zhejiang, China),
and polyclonal rabbit anti-rat phosphorylated (p)-ERK1/2 antibody
(cat. no. 9101S) and polyclonal rabbit anti-rat ERK1/2 antibody
(cat. no. 9102S) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). A UNIQ-10 column Trizol kit was obtained
from Shanghai Sangon Biotech Co., Ltd. (Shanghai, China).
PrimeScript® RT Master Mix Perfect Real Time and
SYBR® Premix Ex Taq™ II were obtained from Biotechnology
Co., Ltd. (Tokyo, Japan).
H9c2 cell culture
H9c2 cells were cultured in Dulbecco's modified
Eagle's medium containing 10% fetal bovine serum (Zhejiang Tianhang
Biological Technology) with 1% penicillin and 1% streptomycin at a
temperature of 37°C. When the cells reached 70–80% confluence, they
were passaged according to a 1:2 proportion and the medium was
changed every 2 days. Cells were seeded into a 35-mm dish at a
density of 1×105. Cells were cultured in serum-free
medium (GE Healthcare Life Sciences, Logan, UT, USA) at 37°C
overnight and pretreated with 100 nM apelin for 2 h, followed by
treatment with 50 ng/ml resistin for 1 h or 48 h.
Determination of cell surface
area
Briefly, 8×104 cells were seeded into a
35-mm dish. Cells were cultured at 37°C with serum-free medium for
18 h and pretreated with 100 nM apelin for 2 h, then treated with
50 ng/ml resistin for 48 h. The cell surface area was measured
using ImageJ version 1.49 software (National Institutes of Health,
Bethesda, MD, USA). Using an inverted microscope, five observation
fields of cells were selected at random and 10 of the cells in each
observation field were selected for measurement of their cell
surface areas (24).
Protein synthesis measurement
Briefly, 1×105 cells were seeded into a
35-mm dish. Cells were cultured with serum-free medium for 18 h and
pretreated with apelin at 100 nM for 2 h, and treated with resistin
for a further 48 h. Cells were digested with 0.25% trypsin
(Beyotime Institute of Biotechnology, Beijing, China) for 1 min and
counted under an inverted microscope. The cells were collected and
lysed with 100 µl tissue lysis buffer (CWBio, Beijing, China).
Protein concentrations were measured using a bicinchoninic acid
(BCA) protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) according to the manufacturer's instructions. Cell protein
synthesis was expressed as the relative protein content, which was
determined by dividing the total protein quantity by the cell
number (24).
Reverse transcription-quantitative
polymerase chain reaction RT-qPCR)
Total RNA was extracted from collected cells using
the UNIQ-10 column TRIzol kit and digested with DNase I. The RNA
concentration was measured with NanoDrop 1000 (Thermo Fisher
Scientific, Waltham, MA, USA). RNA (1 µg) was reverse transcribed
into cDNA using the PrimeScript® RT Master Mix Perfect
Real Time kit. PCR was performed with the SYBR® Premix
Ex Taq™ II kit using an Applied Biosystems® 7500 Fast
Real-Time PCR System (Thermo Fisher Scientific, Inc.). The cycling
conditions for qPCR were as follows: 95°C for 30 sec, and 40 cycles
at 95°C for 5 sec followed by 60°C for 31 sec. 18S rRNA gene
expression served as a control and qPCR data analysis was performed
with the ΔΔCq method (25). The BNP,
β-MHC and 18S primers were designed and synthesized by Shanghai
Sangon Biotech Co., Ltd. and were as follows: Forward,
5′-GGAGCATTGAGTTGGCTCTC-3′ and reverse, 5′-CCAGCTCTCCGAAGTGTTTC-3′
for BNP; forward, 5′-CACCCGCGAGTACAACCTTC-3′ and reverse,
5′-CCCATACCCACCATCACACC-3′ for β-MHC; forward,
5′-CACCCGCGAGTACAACCTTC-3′ and reverse, 5′-CCCATACCCACCATCACACC-3′
for 18S.
Western blot analysis
After the cells reached 80–90% confluence, they were
washed twice with 1X phosphate-buffered saline, digested with 0.25%
trypsin for 1 min and centrifuged at 1,000 × g for 5 min at 4°C.
Cells were added with 100 µl lysis buffer (CWBio) and placed in ice
for 20 min. The lysates were centrifuged at 10,000 × g for 15 min
at 4°C and the supernatant was isolated. The protein concentration
was determined by BCA assay and 5X Laemmli's buffer (CWBio) was
added to samples. The lysates were heated at 95°C for 5 min and
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis. The proteins were transferred onto polyvinylidene
fluoride membranes (Merck Millipore, Billerica, MA, USA) at 200 mA
for 30 min, and blocking was performed with Tris-buffered saline
and Tween-20 (TBST) buffer (20 mM Tris-HCl, 150 mM NaCl and 0.1 %
Tween-20) containing 5% non-fat milk for 1 h at room temperature.
The membranes were incubated in TBST buffer containing 5% non-fat
milk with the following primary antibodies: Polyclonal rabbit
anti-rat p-ERK (1:1,000) and polyclonal rabbit anti-rat ERK
(1:1,000) and polyclonal rabbit anti-rat β-actin (cat. no. 4967S;
1:1,000; Cell Signaling Technology, Inc.) at 4°C overnight. After
incubation with the primary antibodies, horseradish
peroxide-conjugated anti-rabbit secondary antibodies [cat. no.
111-035-003 (polyclonal goat anti-rabbit); 1:10,000; Jackson
ImmunoResearch, Inc., West Grove, PA, USA) were incubated at room
temperature for 1 h. The blots were visualized with an enhanced
chemiluminescence kit (Beijing ComWin Biotech, Beijing, China)
using a FluorChem™ Q Quantitative Western Blot Imaging System
(Bio-Techne, Minneapolis, MN, USA). The densitometry of the bands
was quantified using NIH ImageJ version 1.49 software.
Statistical analysis
All experiments data were expressed as mean ±
standard deviation and performed at least three times. All
statistical analyses were performed by one-way analysis of variance
followed by the Bonferroni post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
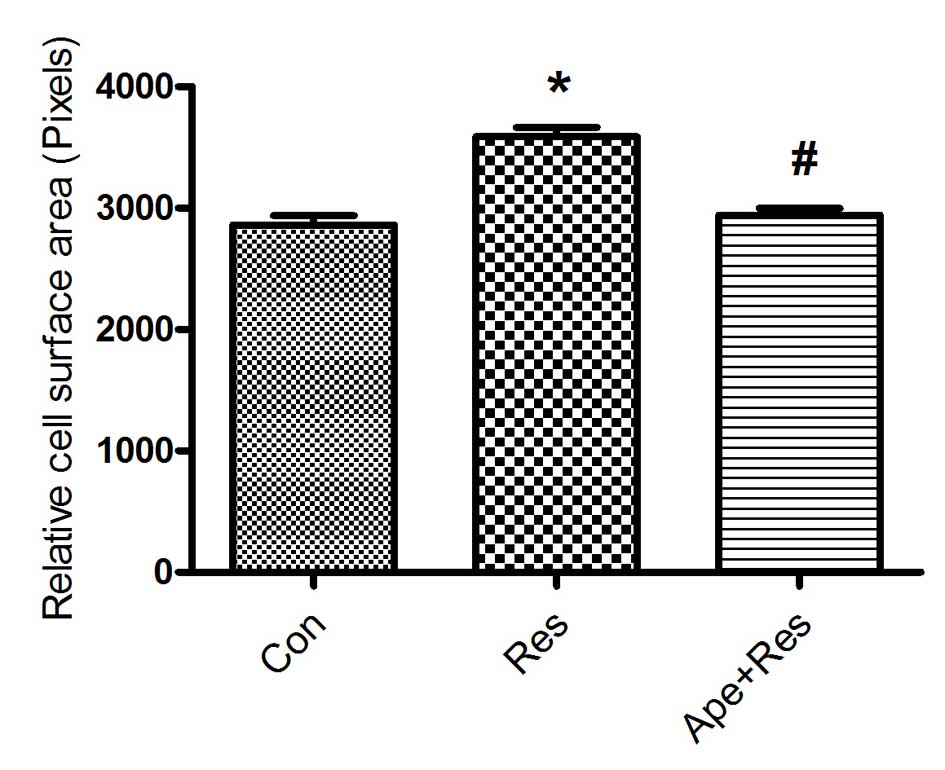
Apelin inhibits resistin-induced H9c2
cell size increase
Resistin treatment was used to induce cardiomyocyte
hypertrophy. H9c2 cells were treated with (50 ng/ml) resistin for
48 h. Resistin significantly increased the cell surface area when
compared with the control group (P<0.01; Fig. 1). Pretreatment of cardiomyocytes with
100 nM apelin, significantly decreased the cell surface area that
had been increased by resistin compared with the resistin group
(P<0.01; Fig. 1).
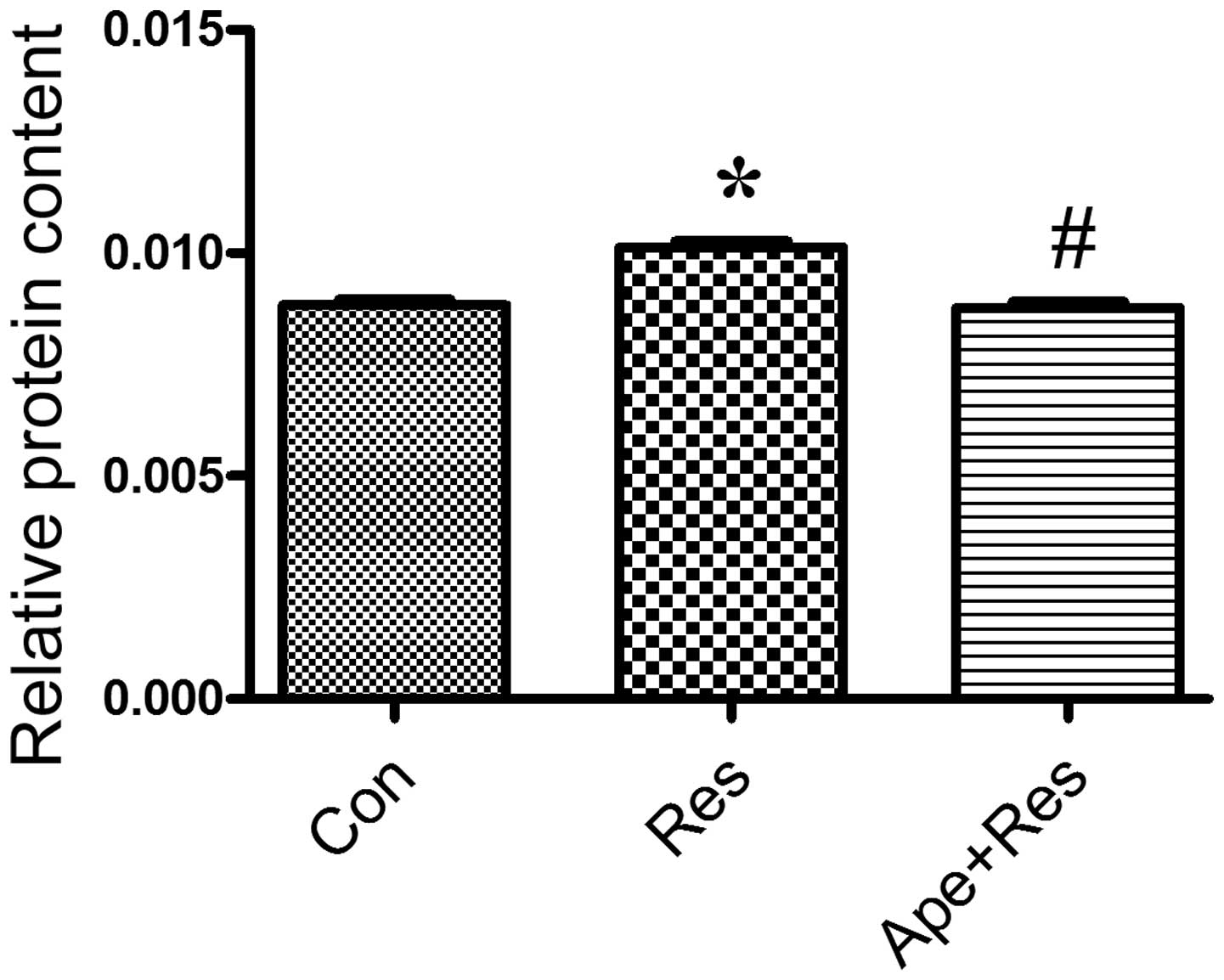
Apelin decreases resistin-induced
cardiomyocyte protein synthesis increase
To analyze whether resistin treatment increases
protein synthesis in H9c2 cells and whether the increase is
inhibited by apelin treatment, cultured cardiomyocytes were exposed
to resistin in the presence and absence of apelin for 48 h.
Resistin significantly increased protein synthesis in
cardiomyocyotes compared with the control group (P<0.01;
Fig. 2). Apelin treatment decreased
the protein synthesis that had been increased by resistin as
compared with the resistin group (P<0.01; Fig. 2).
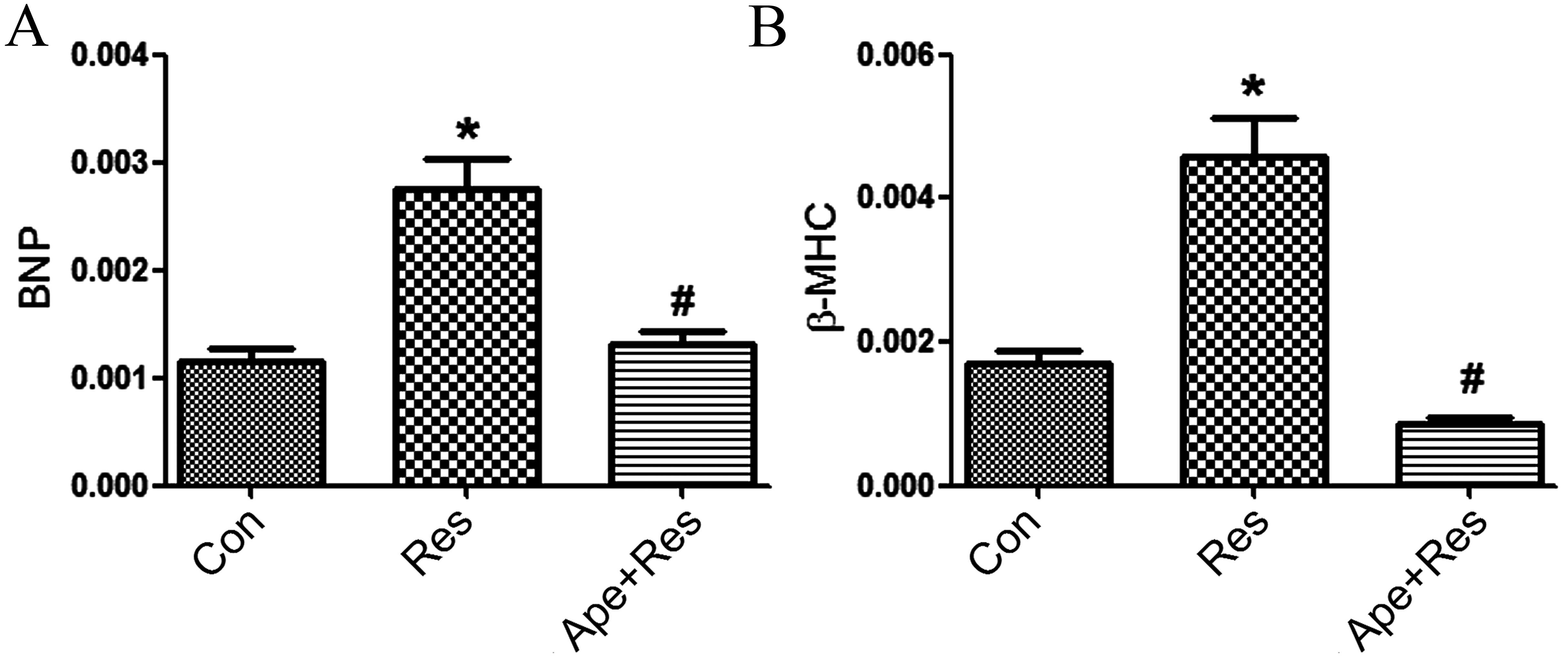
Apelin decreases the resistin-induced
increased expression of BNP and β-MHC mRNA
As BNP and β-MHC are cardiomyocyte hypertrophy
markers, the effect of apelin on expression of BNP and β-MHC mRNA
induced by resistin in H9c2 cells was investigated. Resistin
treatment increased the expression of BNP and β-MHC at the mRNA
level compared with the control group (P<0.01; Fig. 3A and B). Apelin co-treatment with
resistin suppressed resistin-induced increase of BNP and β-MHC mRNA
expression as compared with the resistin group (P<0.01; Fig. 3A and B).
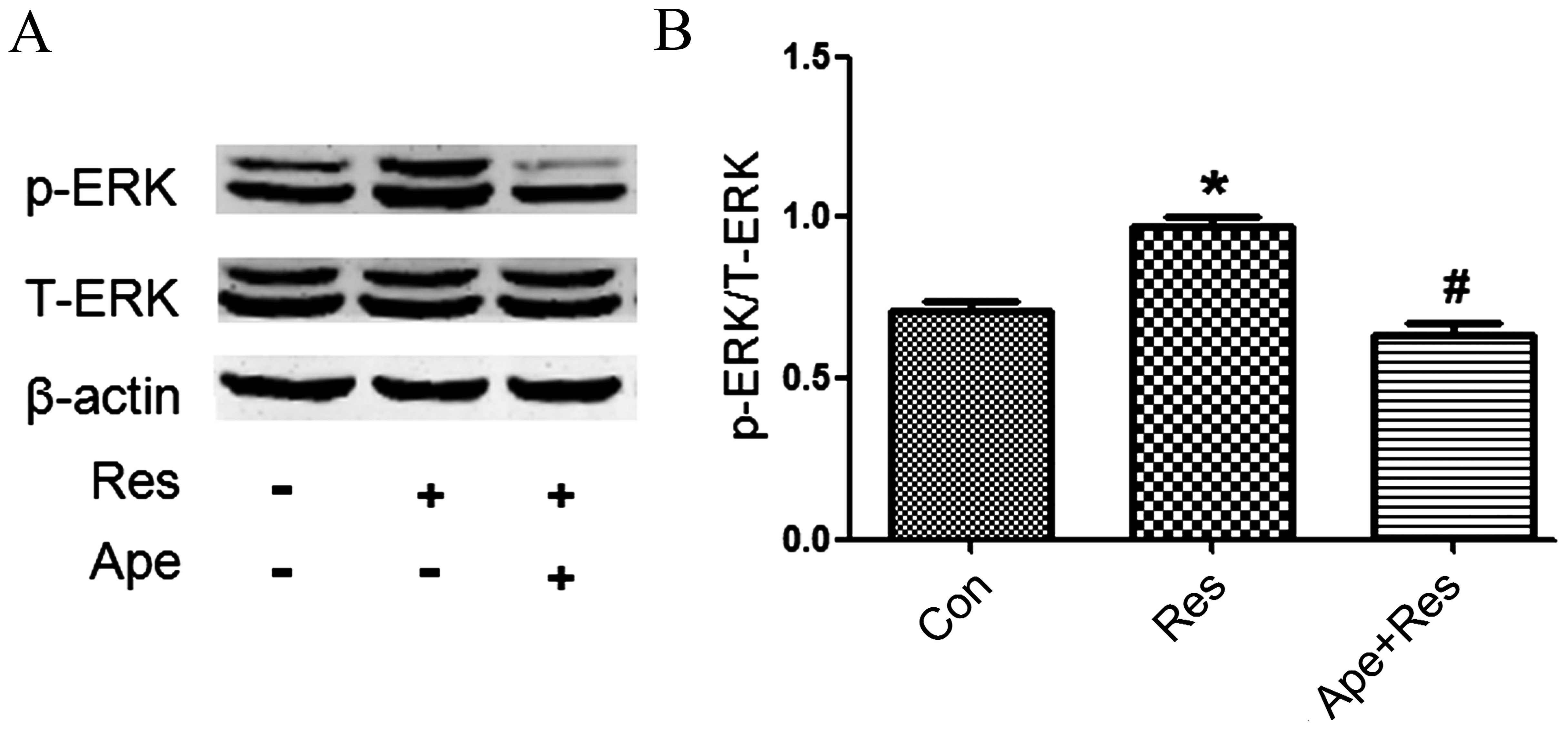
Apelin decreases phosphorylation of
ERK1/2 that is increased by resistin
To elucidate the underlying molecular mechanism by
which apelin inhibits resistin-induced cardiomyocyte hypertrophy,
western blot analysis was performed to detect the phosphorylation
of ERK1/2 upon apelin and resistin co-treatment. Treatment with
resistin decreased phosphorylation of ERK1/2 compared with the
control cells (P<0.01; Fig. 4),
whereas total ERK1/2 protein expression remained unchanged. By
contrast, pretreatment of apelin suppressed expression of p-ERK1/2
as compared with the resistin group (P<0.01; Fig. 4).
Discussion
Previous studies have indicated that resistin
induces cardiac hypertrophy (8), while
apelin inhibits cardiac hypertrophy (23); however, the underlying molecular
mechanisms by which apelin inhibits resistin-induced cardiac
hypertrophy remain largely unknown. To the best of our knowledge,
this is the first study to investigate apelin suppressing
resistin-induced cardiomyocyte hypertrophy via the inactivation of
the ERK1/2 signaling pathway. In the current study, resistin
increased cell size, protein synthesis and the expression of
hypertrophic markers, BNP and β-MHC at the mRNA level, whereas
apelin suppressed these effects that were induced by resistin. This
indicated that resistin-induced cardiomyocyte hypertrophy may be
inhibited by apelin.
Resistin is a secreted adipokine. Resistin function
is associated with obesity, diabetes and insulin resistance.
Treatment with resistin impairs glucose tolerance and insulin
action, whereas loss of resistin function improves insulin
resistance (9,26,27) and
Resistin promotes endothelial dysfunction and I/R myocardial injury
(28–30). Hyperresistinemia may contribute to the
impairment of cardiac contractility and diabetic cardiac function
(31). Overexpression of resistin in
vivo using adeno-associated virus serotype 9 significantly
decreases left ventricular contractility and induces oxidative
stress, fibrosis, apoptosis and myocardial remodeling in normal
rats (32). Furthermore,
overexpression of resistin induces cardiac hypertrophy in neonatal
rat cardiomyocytes through activation of oxidative stress, insulin
receptor substrate 1 (IRS1)/mitogen-activated protein kinase (MAPK)
(8), AMPK/mechanistic target of
rapamycin/p70S6 kinase and apoptosis signal-regulating kinase
1/c-Jun N-terminal kinases/IRS1 signaling pathways (33). In the current study, H9c2 cells were
used as a model and treated with resistin to induce cardiomyocyte
hypertrophy. Treatment with resistin also induced an increase in
ERK1/2 phosphorylation, indicating that resistin induces
cardiomyocyte hypertrophy via activation of the ERK signaling
pathway.
The adipokine, apelin is an endogenous ligand for
the G protein-coupled receptor APJ. Apelin and APJ are expressed in
the heart. Adipocytes secrete apelin and cardiomyocytes also
secrete apelin (34). Human studies
have demonstrated that the apelin-AJP system is downregulated in
the hypertrophic heart (35,36). Apelin treatment abolishes development
of cardiac hypertrophy, as well as preventing fibrosis progression
and cardiac contractile dysfunction (37). In addition, apelin gene therapy
increases myocardial vascular density and ameliorates diabetic
cardiomyopathy via upregulation of sirtuin 3 (38). Previous studies demonstrated that
apelin treatment contributes to cardioprotection in cardiac I/R
injury, as well as angiotensin II- or isoproterenol-induced cardiac
remodeling (39,40). In the absence of apelin, stretch
signals through the apelin receptor are mediated via β-arrestins
resulting in detrimental cardiac hypertrophy (41). Notably, apelin knockout mice display
impaired cardiac contractility with aging and developed progressive
heart failure induced by pressure overload (19). Furthermore, apelin knockout mice and
APJ knockout mice showed only modest declines in cardiac function
(42). It has been demonstrated that
ERK1/2 signaling is necessary for promoting hypertrophic growth.
ERK1 and ERK2 are regulated by MAPK kinases (MEK) 1 and MEK2, and
ERK1/2 proteins are phosphorylated by MEK1/2 at a threonine and
adjacent tyrosine residue. The ERK1/2 signaling pathway has been
associated with the development of cardiac hypertrophy and cardiac
failure (43). The present study
demonstrated that resistin increases phosphorylation of ERK1/2,
whereas apelin decreases the phosphorylation of ERK1/2, which was
increased by resistin. These results indicate that resistin-induced
cardiomyocyte hypertrophy is inhibited by apelin via inactivation
of the ERK1/2 signaling pathway.
In conclusion, resistin exposure causes increased
BNP and β-MHC mRNA expression levels, greater cell surface area and
protein synthesis, as well as increased ERK1/2 phosphorylation,
while apelin inhibits these resistin-induced effects. These
findings indicate that apelin inhibits resistin-induced
cardiomyocyte hypertrophy via inactivation of the ERK1/2 signaling
pathway. The present results provide novel insight, presenting
apelin as a useful treatment for cardiac hypertrophy.
Acknowledgements
The present study was supported by the Key Research
and Development Program of Shaanxi Province (grant no.
201603D321057) and Basic and Cutting-Edge Technology Research
Project of Henan Province (grant no. 142300410118).
References
|
1
|
Maron BJ: Hypertrophic cardiomyopathy: A
systematic review. JAMA. 287:1308–1320. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chugh SS, Reinier K, Teodorescu C, Evanado
A, Kehr E, Al Samara M, Mariani R, Gunson K and Jui J: Epidemiology
of sudden cardiac death: Clinical and research implications. Prog
Cardiovasc Dis. 51:213–228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beohar N, Zajarias A, Thourani VH,
Herrmann HC, Mack M, Kapadia S, Green P, Arnold SV, Cohen DJ,
Généreux P, et al: Analysis of early out-of hospital mortality
after transcatheter aortic valve implantation among patients with
aortic stenosis successfully discharged from the hospital and alive
at 30 days (from the placement of aortic transcatheter valves
trial). Am J Cardiol. 114:1550–1555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeong K, Kwon H, Min C and Pak Y:
Modulation of the caveolin-3 localization to caveolae and STAT3 to
mitochondria by catecholamine-induced cardiac hypertrophy in H9c2
cardiomyoblasts. Exp Mol Med. 41:226–235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee H, Yoo YS, Lee D and Song EJ:
Cholesterol induces cardiac hypertrophy by activating the AKT
pathway. J Steroid Biochem Mol Biol. 138:307–313. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steppan CM, Bailey ST, Bhat S, Brown EJ,
Banerjee RR, Wright CM, Patel HR, Ahima RS and Lazar MA: The
hormone resistin links obesity to diabetes. Nature. 409:307–312.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim M, Oh JK, Sakata S, Liang I, Park W,
Hajjar RJ and Lebeche D: Role of resistin in cardiac contractility
and hypertrophy. J Mol Cell Cardiol. 45:270–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Muse ED, Obici S, Bhanot S, Monia BP,
McKay RA, Rajala MW, Scherer PE and Rossetti L: Role of resistin in
diet-induced hepatic insulin resistance. J Clin Invest.
114:232–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Graveleau C, Zaha VG, Mohajer A, Banerjee
RR, Dudley-Rucker N, Steppan CM, Rajala MW, Scherer PE, Ahima RS,
Lazar MA, et al: Mouse and human resistins impair glucose transport
in primary mouse cardiomyocytes, and oligomerization is required
for this biological action. J Biol Chem. 280:31679–31685. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen YH, Hung PF and Kao YH: IGF-I
downregulates resistin gene expression and protein secretion. Am J
Physiol Endocrinol Metab. 288:E1019–E1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patel L, Buckels AC, Kinghorn IJ, Murdock
PR, Holbrook JD, Plumpton C, Macphee CH and Smith SA: Resistin is
expressed in human macrophages and directly regulated by PPAR gamma
activators. Biochem Biophys Res Commun. 300:472–476. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Steppan CM and Lazar MA: The current
biology of resistin. J Intern Med. 255:439–447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pang SS and Le YY: Role of resistin in
inflammation and inflammation-related diseases. Cell Mol Immunol.
3:29–34. 2006.PubMed/NCBI
|
|
15
|
Bokarewa M, Nagaev I, Dahlberg L, Smith U
and Tarkowski A: Resistin, an adipokine with potent proinflammatory
properties. J Immunol. 174:5789–5795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kunnari AM, Savolainen ER, Ukkola OH,
Kesäniemi YA and Jokela MA: The expression of human resistin in
different leucocyte lineages is modulated by LPS and TNFalpha.
Regul Pept. 157:57–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schwartz DR and Lazar MA: Human resistin:
Found in translation from mouse to man. Trends Endocrinol Metab.
22:259–265. 2011.PubMed/NCBI
|
|
18
|
Szokodi I, Tavi P, Földes G,
Voutilainen-Myllylä S, Ilves M, Tokola H, Pikkarainen S, Piuhola J,
Rysä J, Tóth M, et al: Apelin, the novel endogenous ligand of the
orphan receptor APJ, regulates cardiac contractility. Circ Res.
91:434–440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuba K, Zhang L, Imai Y, Arab S, Chen M,
Maekawa Y, Leschnik M, Leibbrandt A, Markovic M, Schwaighofer J, et
al: Impaired heart contractility in Apelin gene-deficient mice
associated with aging and pressure overload. Circ Res. 101:e32–e42.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maguire JJ, Kleinz MJ, Pitkin SL and
Davenport AP: [Pyr1]apelin-13 identified as the predominant apelin
isoform in the human heart: Vasoactive mechanisms and inotropic
action in disease. Hypertension. 54:598–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang W, McKinnie SM, Patel VB, Haddad G,
Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, et al:
Loss of Apelin exacerbates myocardial infarction adverse remodeling
and ischemia-reperfusion injury: Therapeutic potential of synthetic
Apelin analogues. J Am Heart Assoc. 2:e0002492013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Zeng H and Chen JX: Apelin-13
increases myocardial progenitor cells and improves repair
postmyocardial infarction. Am J Physiol Heart Circ Physiol.
303:H605–H618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ceylan-Isik AF, Kandadi MR, Xu X, Hua Y,
Chicco AJ, Ren J and Nair S: Apelin administration ameliorates high
fat diet-induced cardiac hypertrophy and contractile dysfunction. J
Mol Cell Cardiol. 63:4–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu P, Cheng GC, Ye QH, Deng YZ and Wu L:
LKB1/AMPK pathway mediates resistin-induced cardiomyocyte
hypertrophy in H9c2 embryonic rat cardiomyocytes. Biomed Rep.
4:387–391. 2016.PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banerjee RR, Rangwala SM, Shapiro JS, Rich
AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, et al:
Regulation of fasted blood glucose by resistin. Science.
303:1195–1198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rajala MW, Obici S, Scherer PE and
Rossetti L: Adipose-derived resistin and gut-derived resistin-like
molecule-beta selectively impair insulin action on glucose
production. J Clin Invest. 111:225–230. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao J, Chua C Chang, Chen Z, Wang H, Xu X,
C Hamdy R, McMullen JR, Shioi T, Izumo S and Chua BH: Resistin,
anadipocytokine, offers protection against acute myocardial
infarction. J Mol Cell Cardiol. 43:601–609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gualillo O, González-Juanatey JR and Lago
F: The emerging role of adipokines as mediators of cardiovascular
function: Physiologic and clinical perspectives. Trends Cardiovasc
Med. 17:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rothwell SE, Richards AM and Pemberton CJ:
Resistin worsens cardiac ischaemia-reperfusion injury. Biochem
Biophys Res Commun. 349:400–407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bobbert P, Jenke A, Bobbert T, Kühl U,
Rauch U, Lassner D, Scheibenbogen C, Poller W, Schultheiss HP and
Skurk C: High leptin and resistin expression in chronic heart
failure: Adverse outcome in patients with dilated and inflammatory
cardiomyopathy. Eur J Heart Fail. 14:1265–1275. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chemaly ER, Hadri L, Zhang S, Kim M,
Kohlbrenner E, Sheng J, Liang L, Chen J, K-Raman P, Hajjar RJ, et
al: Long-term in vivo resistin overexpression induces myocardial
dysfunction and remodeling in rats. J Mol Cell Cardiol. 51:144–155.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kang S, Chemaly ER, Hajjar RJ and Lebeche
D: Resistin promotes cardiac hypertrophy via the AMP-activated
protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun
N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways.
J Biol Chem. 286:18465–18473. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kageyama K, Hanada K and Suda T:
Regulation of corticotropin-releasing factor receptor type 2beta
mRNA by mitogen-activated protein kinases in aortic smooth muscle
cells. Regul Pept. 126:223–231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iwanaga Y, Kihara Y, Takenaka H and Kita
T: Down-regulation of cardiac apelin system in hypertrophied and
failing hearts: Possible role of angiotensin II-angiotensin type 1
receptor system. J Mol Cell Cardiol. 41:798–806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Földes G, Horkay F, Szokodi I, Vuolteenaho
O, Ilves M, Lindstedt KA, Mäyränpää M, Sármán B, Seres L, Skoumal
R, et al: Circulating and cardiac levels of apelin, the novel
ligand of the orphan receptor APJ, in patients with heart failure.
Biochem Biophys Res Commun. 308:480–485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pchejetski D, Foussal C, Alfarano C,
Lairez O, Calise D, Guilbeau-Frugier C, Schaak S, Seguelas MH,
Wanecq E, Valet P, et al: Apelin prevents cardiac fibroblast
activation and collagen production through inhibition of
sphingosine kinase 1. Eur Heart J. 33:2360–2369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zeng H, He X, Hou X, Li L and Chen JX:
Apelin gene therapy increases myocardial vascular density and
ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3.
Am J Physiol Heart Circ Physiol. 306:H585–H597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jia YX, Pan CS, Zhang J, Geng B, Zhao J,
Gerns H, Yang J, Chang JK, Tang CS and Qi YF: Apelin protects
myocardial injury induced by isoproterenol in rats. Regul Pept.
133:147–154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Siddiquee K, Hampton J, Khan S, Zadory D,
Gleaves L, Vaughan DE and Smith LH: Apelin protects against
angiotensin II-induced cardiovascular fibrosis and decreases
plasminogen activator inhibitor type-1 production. J Hypertens.
29:724–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Scimia MC, Hurtado C, Ray S, Metzler S,
Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, et al:
APJ acts as a dual receptor in cardiac hypertrophy. Nature.
488:394–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Charo DN, Ho M, Fajardo G, Kawana M, Kundu
RK, Sheikh AY, Finsterbach TP, Leeper NJ, Ernst KV, Chen MM, et al:
Endogenous regulation of cardiovascular function by apelin-APJ. Am
J Physiol Heart Circ Physiol. 297:H1904–H1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mutlak M and Kehat I: Extracellular
signal-regulated kinases 1/2 as regulators of cardiac hypertrophy.
Front Pharmacol. 6:1492015. View Article : Google Scholar : PubMed/NCBI
|