Introduction
Chronic kidney disease (CKD) contributes
significantly to the global health burden; it has an estimated
prevalence of 8–16% worldwide and results in millions of
mortalities annually, due to poor access to affordable treatment in
resource-limited settings (1,2). CKD is an important determinant of the
poor health outcomes of major non-communicable diseases, and is a
notable risk multiplier in patients with diabetes and hypertension
(3). There is a paucity of data
regarding the epidemiology of the early stages of pediatric CKD, as
a result of under-diagnosis and under-reporting. However,
statistics indicate that congenital disorders, including congenital
anomalies of the kidney and urinary tract, and hereditary
nephropathies, are responsible for approximately 2/3 of all cases
of CKD in developed countries, whereas acquired causes predominate
in developing countries (4). There are
very clear geographic diversities in the reported causes of
pediatric CKD, which are attributed to dissimilarities in clime,
race, hereditary and ancestry (5).
Thus, familial clustering and disparities in CKD prevalence rates
across ethnic and racial groups indicate that progression of renal
disease has a strong genetic component (6–9).
Previous hereditary-associated studies have revealed
common loci that may increase CKD risk (10). However, the majority of risk alleles
for CKD identified by the genome-wide association studies (GWAS)
confer only a very small relative risk. For example, the CKD
Genetics study groups conducted a meta-analysis of GWAS data in
67,093 subjects of Caucasian descent from 20 predominantly
demographic studies; the 16 analyzed loci accounted for only 1.4%
of the variation in estimated glomerular filtration rate,
indicating that other hereditary and non-genetic factors may be
associated with CKD risk (10).
According to this concept of ‘missing heritability’, epigenetic
modifications presumably contribute to the inherited risk for CKD,
as well as contribute to the explanation for the environmental
influence on the human genome, which alters an individual's
susceptibility to disease. Although there is no change in DNA
sequence, modification in genome structure results in heritable and
potentially reversible alterations in gene expression (11). Notably, mammalian studies have
demonstrated a feasible link between nutrition and non-genetic
exposure, around the time of conception, as well as in epigenetic
changes, in the expression of major genes identified in renal
organogenesis (11). The major
consequence is the reduction in the number of nephrons, with
subsequent predisposition to hypertension and CKD. Similarly,
adverse intrauterine and postnatal conditions have a prolonged role
in the evolution of CKD over the lifetime of an individual. Such
adverse intrauterine conditions include small-for-gestational age
infants or conversely, macrosomic infants of diabetic mothers. In
addition, poor diabetes control ≥25 years earlier leads to
increased susceptibility to nephropathy regardless of a decade of
excellent glycemic control (the ‘metabolic memory’ phenomenon).
Identifying these epigenetic changes is crucial because of their
potentially reversible nature; these epigenetic changes may serve
as future therapeutic targets to prevent kidney fibrosis and CKD
(11). Despite the increase in
epigenetic research and the rising evidence of its relevance in the
evolution of various complex disease traits, it is more prominent
in oncology. Although the deviation into epigenetics by other
subspecialties remains tentative, such research is largely
experimental with few existing reports regarding the clinical
implication of epigenetics in kidney disease (12). The aim of the current review was to
appraise the role of epigenetics in CKD, and highlight potential
future therapeutic pathways.
Epigenetics: Definition and
pathophysiology
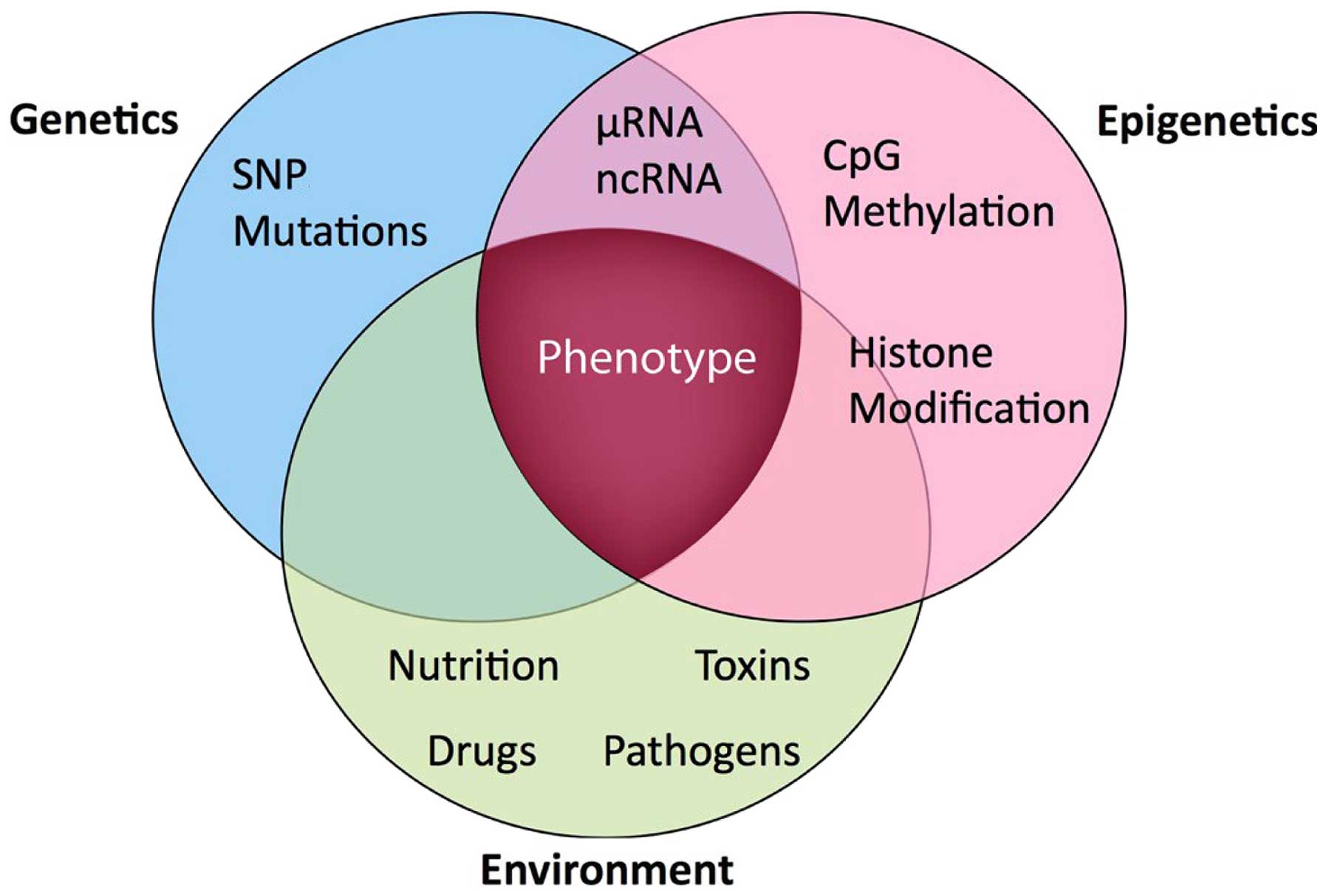
The term ‘epigenetics’ is best understood within the
context of the interaction between the gene and the environment
(Fig. 1). From a historical
perspective, the definition of the term has metamorphosed over the
years from its original coinage and meaning by Slack (13). More than 10 years later, another author
defined ‘epigenetic systems’ as supportive processes involved in
ascertaining the specific genes that will be expressed in any
individual cell (14). Following the
identification of heritable models of DNA methylation, the
hypothesis that epigenetic characteristics were inherited as
controlling indicators coupled with the genome predominated.
Epigenetics was subsequently defined as the investigation of
mitotically and/or meiotically inheritable alterations in gene
expression, which are not associated with alterations in DNA
sequence (15). The identification of
the regulatory function of histone post-translational modifications
and their reciprocal association with transcriptional states
encouraged a more relaxed use of the term ‘epigenetics’, to refer
to any molecular imprint located on chromosomes (particularly
histone marks) and loosely defined as the configurational
adjustment of chromosomal loci in order to express, indicate or
prolong altered activity (16).
Simultaneously, a narrower definition was suggested as a
consistently inheritable phenotype due to changes in a chromosome,
which are not associated with alterations in the DNA sequence
(17). Subsequently, epigenetics came
to be defined as the inheritance of variation (−genetics) above and
beyond (epi-) alterations in the DNA sequence (18). Therefore, three main epigenetic
mechanisms are recognized in mammals as follows: Changes in DNA
methylation, histone post-translational modifications and RNA
interference (non-coding RNAs) (19).
DNA methylation is an important epigenetic factor that is
responsible for the control of gene expression and genomic
consistency, and is biologically vital for the preservation of
various physiologic activities of the cell (20).
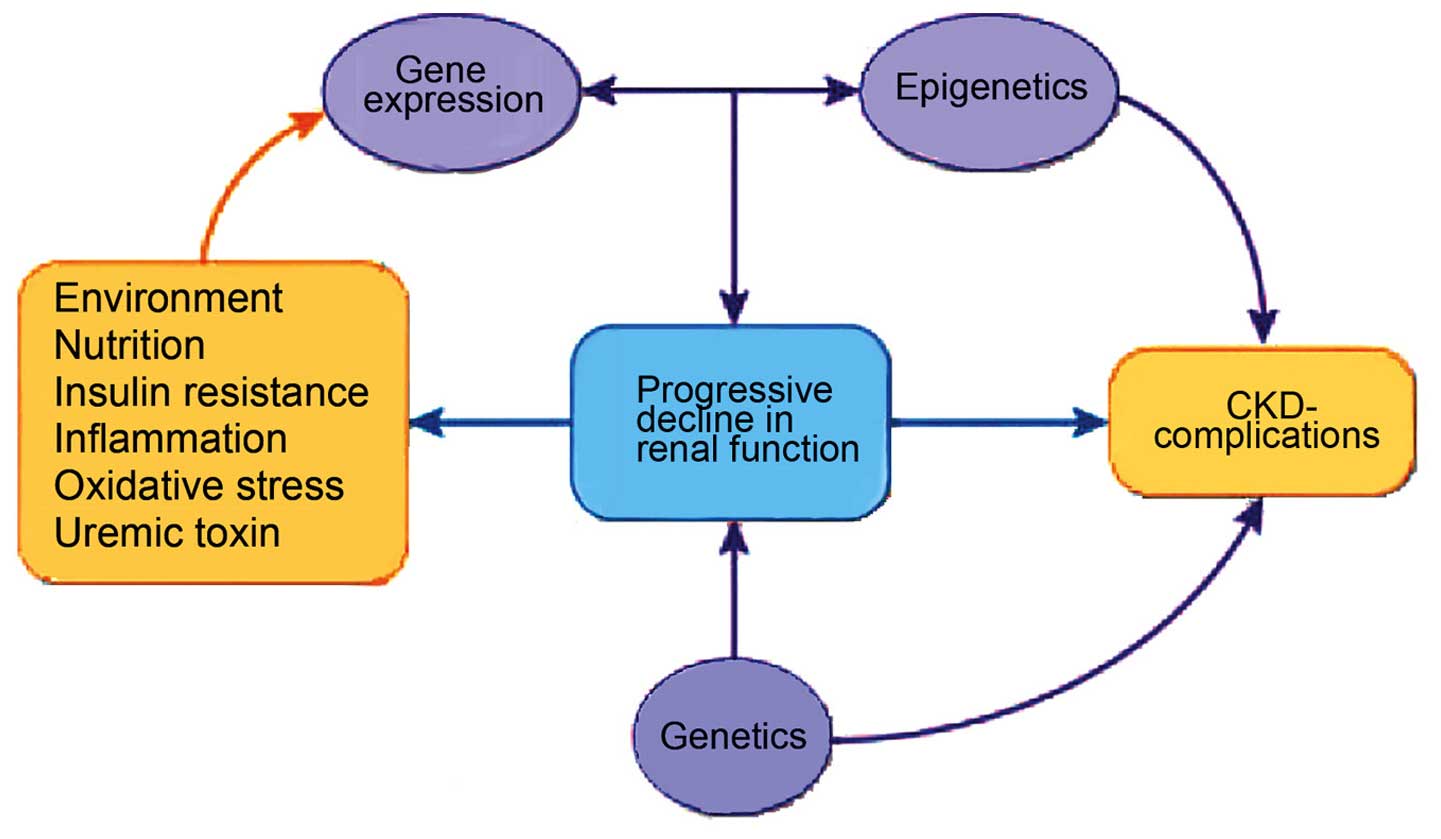
The epigenome is the interconnecting point of
genetics and environment, which stimulates changes that modulate
disease phenotype by direct effect on the target gene, irrespective
of changes in sequence within the gene (Fig. 2). Possible triggers include
environmental signals and pathologic states, such as diet,
exercise, inflammation, oxidative stress, metabolic changes and
toxins (21–23). Epigenetic changes are potentially
reversible (24,25); notably, to simple measures, such as
lifestyle modifications and nutrient supplementation (26–29).
Regarding the pathophysiology of epigenetic
processes, DNA methylation occurs at the 5-cystine of CpG
dinucleotides. Briefly, hypermethylation switches off the gene
expression (gene silencing), while hypomethylation switches it on
or leads to the re-expression of a normally silenced gene.
Hypomethylation and genomic disequilibrium results from imbalances
in dietary nutrients, such as folate (29). Methyl groups originating from the diet
are passed on to DNA via folate and methionine pathways, and
5-methyl cytosine content is modified by the nutritional
accessibility to folate (19).
Furthermore, infections (particularly viral infections) initiate
DNA methylation. Thus, variations in DNA-methylation patterns occur
following dietary changes, inherited genetic polymorphisms and
exposure to environmental factors.
Additionally, with respect to histone modification,
DNA is transcriptionally active when histones are acetylated and
unmethylated, whereas deacetylated and methylated histones block
gene expression (30). Histone
methylation occurs on Arg or Lys residues, with a discriminating
site selectivity for Lys methylation at specific positions in the
N-end points of histones H3 and H4. Thus, Lys methylation is added
to acetylation and phosphorylation as a third component of a
‘histone code’, which alters the fundamental chromatin makeup of
the genome. The adjustment of this ‘histone code’ influences the
maintenance of gene expression with inheritable epigenetic patterns
(31).
Finally, cleavage and degradation of microRNA
(miRNA) block translational mechanisms and prevent protein
formation, resulting in silencing of gene expression.
Double-stranded RNAs also cause transcriptional silencing via
methylation of homologous DNA promoter sequences (32) and/or formation of heterochromatin
(33). This RNA interference is vital
for the evolution of kidney disease (12), as miRNAs are critical in the
preservation of glomerular balance (34).
Role of epigenetics in CKD
A recent review highlights various evidence-based
studies, which show the association between the intrauterine
environment/number of nephrons, and the development of nephropathy
(35). For example, epidemiologic data
has revealed an association between low birth weight and subsequent
non-communicable diseases, such as adult hypertension, diabetes,
cardiovascular disease and CKD (36).
Furthermore, a direct correlation between low birth weight and
microalbuminuria has been reported in insulin-dependent diabetes
mellitus (IDDM), as well as in older non-diabetic subjects
(37). Low birth weight also directly
correlates with albuminuria in non-IDDM and with the progression of
CKD (37). Notably, findings from
animal and human studies support the hypothesis that low birth
weight (a reflection of an unfavorable intrauterine environment) is
accompanied by a reduction in the number of nephrons at birth
(37). In man, the ultimate
acquisition of nephrons depends on the intrauterine milieu and
gestational age at delivery, as post-natal nephrogenesis is absent.
Growth-retarded stillbirths and live-born infants with intrauterine
growth retardation demonstrated a diminished number of nephrons
when compared with control infants whose birth weights were
appropriate for their gestational age (38). However, the caveat is that normal birth
weight is not often synonymous with a conventional number of
nephrons. To further support these observations, autopsy studies in
adults indicated a significant correlation between birth
weights/number of nephrons, and mean arterial pressure (39). A previous study described a case series
(n=6) of focal segmental glomerulosclerosis (FSGS) where the
clinicopathologic results strongly agreed with the secondary form
of FSGS, in which the only observed risk factors were prematurity
and very low birth weight (40).
There is substantial evidence showing the role of
key epigenetic mechanisms in the progression of CKD. A previous
study observed a difference in DNA methylation patterns identified
in renal fibroblasts between normal and diseased kidneys (41); DNA methylation may regulate fibroblast
multiplication and contribute to renal fibrogenesis. For example,
in the study, experimental murine models treated with
5-azacytidine, in which DNA methylation was impeded, experienced
protection from kidney fibrosis. The observation further
underscores the fact that hypermethylation contributes to renal
fibrogenesis, and is the first report to show that DNA methylation
directly affects the evolution of CKD. In addition, these
investigators also demonstrated that hypermethylation of RAS
protein activator like-1 was associated with the sustenance of
fibroblast proliferation and renal fibrogenesis (41).
Although the various histone modifications in the
human kidney have not been evaluated with respect to CKD, studies
conducted in various murine models of nephropathy indicate major
disparities in the ‘histone code’ (35). Certain studies demonstrated that
elevated levels of renal H3K9 and H3K23 acetylation, H3K4
dimethylation and H3 phosphorylation at serine 10 were noted in
murine models of advanced diabetic nephropathy (42,43).
Furthermore, there are experimental instances showing the effect of
posttranslational histone modifications, which induce renin cell
identity. Using a cultured renin linage cell system, studies
observed that its gene expression memory is maintained in the
cultured cells and may be re-enacted by cAMP and chromatin
remodeling (histone H4 acetylation) (44). Other investigators also noted that
CREB-binding protein and p300 (which possess histone
acetyltransferase activity) are vital for renin cell
distinctiveness, as well as the integrity of the renal architecture
(45).
Finally, the evidence for the role of RNA
interference in CKD includes the following: First, one study
demonstrated that after dicer (miRNA-generating enzyme) was
inactivated in murine podocytes, the experimental mice presented
with proteinuria and subsequently succumbed due to renal failure
(46). The glomeruli have demonstrated
effacement of the foot process, podocyte cell death, expansion of
the mesangium and glomerulosclerosis (47). Additionally, previous studies have
identified that disruption of miRNA biogenesis in murine podocytes
was followed by proteinuria, podocyte dedifferentiation and
crescent structure resulting in end-stage renal disease (ESRD)
(48,49).
Future therapeutic trajectories
Control of the epigenetic gene is hypothetically
compliant to interventional measures given the non-mutation of the
gene by methylation and the fact that the chromatin is not
irreversibly changed (12). An
interventional study evaluated the outcome of folate administration
on DNA methylation in ESRD subjects with hyperhomocysteinaemia
(50); the preliminary findings
support a previous observation that hyperhomocysteinaemia-induced
DNA hypomethylation could be reversed by administration of folate
(51).
Notably, numerous epigenetic therapeutic agents are
in various developmental phases despite their limitation in target
specificity (52). Certain epigenetic
therapeutic agents include nucleoside and non-nucleoside analogs,
which are also known as inhibitors of DNA methylation (52). For example, myelodysplastic syndrome
and secondary acute myeloid leukemia, which are unresponsive to
conventional chemotherapy, have been managed with DNA
methyltransferase inhibitors, including 5-azacytidine and
decitabine with favorable outcomes (53).
Furthermore, inhibitors of histone deacytylase,
which consist of a large, heterogeneous group of therapeutic agents
comprising short-chain fatty acids, hydroxamic acids, benzamides
and cyclic tetrapeptides are also within the future treatment
trajectory for CKD. Specifically, all trans-retinoic acids and
13-cis-retinoic acid are triggers of hyperacetylation. In addition,
they are being used experimentally in the treatment of pauci-immune
vasculitis (12). On a theoretical
basis, small interfering RNA may be employed to treat any disease
arising from overexpression of a distinctive gene. Therefore, this
technique might be applicable to genes implicated in inflammation,
proliferation and fibrosis. In fact, nucleic acid-based
interventions are currently being performed for atherosclerosis and
progressive renal diseases (54). As
there are more breakthroughs regarding the role of epigenetics in
CKD, it is hoped that novel markers for diagnostic purposes, as
well as treatment-driven molecular pathways will continue to emerge
in the field of renal medicine.
Conclusion
Advances in comprehending epigenetic alterations in
the evolution of CKDs may significantly predict the speed of
disease progression, develop targeted treatment strategies in
preventing its progression, and provide an efficient management of
uremia-associated complications (12).
The current limitation of epigenetic therapeutic agents, which
remain largely in the developmental stage, is their lack of target
specificity. Furthermore, there are ongoing efforts to conduct gene
expression studies and epigenomics analysis to identify novel
diagnostic and prognostic markers for progressive renal diseases.
Therapeutic trajectories for CKD in children based on the influence
of epigenetics may in future revolutionize the management of
CKD.
References
|
1
|
World Kidney Day, . Chronic Kidney
Disease. 2015.http://www.worldkidneyday.org/faqs/chronic-kidney-disease/Accessed
October 10, 2014.
|
|
2
|
Jha V, Garcia-Garcia G, Iseki K, Li Z,
Naicker S, Plattner B, Saran R, Wang AY and Yang CW: Chronic kidney
disease: Global dimension and perspectives. Lancet. 382:260–272.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Couser WG, Remuzzi G, Mendis S and Tonelli
M: The contribution of chronic kidney disease to the global burden
of major noncommunicable diseases. Kidney Int. 80:1258–1270. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Harambat J, van Stralen KJ, Kim JJ and
Tizard EJ: Epidemiology of chronic kidney disease in children.
Pediatr Nephrol. 27:363–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warady BA and Chadha V: Chronic kidney
disease in children: The global perspective. Pediatr Nephrol.
22:1999–2009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bowden DW: Genetics of kidney disease.
Kidney Int Suppl. 83:S8–S12. 2003. View Article : Google Scholar
|
|
7
|
Lei HH, Perneger TV, Klag MJ, Whelton PK
and Coresh J: Familial aggregation of renal disease in a
population-based case-control study. J Am Soc Nephrol. 9:1270–1276.
1998.PubMed/NCBI
|
|
8
|
Hsu CY, Lin F, Vittinghoff E and Shlipak
MG: Racial differences in the progression from chronic renal
insufficiency to end-stage renal disease in the United States. J Am
Soc Nephrol. 14:2902–2907. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Freedman BI, Spray BJ, Tuttle AB and
Buckalew VM Jr: The familial risk of end-stage renal disease in
African Americans. Am J Kidney Dis. 21:387–393. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Köttgen A, Pattaro C, Böger CA,
Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith
AV, et al: New loci associated with kidney function and chronic
kidney disease. Nat Genet. 42:376–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liakopoulos V, Georgianos PI,
Eleftheriadis T and Sarafidis PA: Epigenetic mechanisms and kidney
diseases. Curr Med Chem. 18:1733–1739. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dwivedi RS, Herman JG, McCaffrey TA and
Raj DSC: Beyond genetics: Epigenetic code in chronic kidney
disease. Kidney Int. 79:23–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slack JM: Conrad Hal Waddington: The last
Renaissance biologist? Nat Rev Genet. 3:889–895. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nanney DL: Epigenetic control systems.
Proc Natl Acad Sci USA. 44:712–717. 1958. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riggs A, Martienssen R and Russo V:
Epigenetic mechanisms of gene regulation. 32. Cold Spring Harbor
Laboratory Press; 1996
|
|
16
|
Bird A: Perceptions of epigenetics.
Nature. 447:396–398. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berger SL, Kouzarides T, Shiekhattar R and
Shilatifard A: An operational definition of epigenetics. Genes Dev.
23:781–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonasio R, Tu S and Reinberg D: Molecular
signals of epigenetic states. Science. 330:612–616. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Link A, Balaguer F and Goel A: Cancer
chemoprevention by dietary polyphenols: Promising role for
epigenetics. Biochem Pharmacol. 80:1771–1792. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sadikovic B, Al-Romaih K, Squire JA and
Zielenska M: Cause and consequences of genetic and epigenetic
alterations in human cancer. Curr Genomics. 9:394–408. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luch A: Nature and nurture-lessons from
chemical carcinogenesis. Nat Rev Cancer. 5:113–125. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perna AF, Ingrosso D, Galletti P, Zappia V
and De Santo NG: Membrane protein damage and methylation reactions
in chronic renal failure. Kidney Int. 50:358–366. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robertson KD and Wolffe AP: DNA
methylation in health and disease. Nat Rev Genet. 1:11–19. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ingrosso D, Cimmino A, Perna AF, Masella
L, De Santo NG, De Bonis ML, Vacca M, D'Esposito M, D'Urso M,
Galletti P, et al: Folate treatment and unbalanced methylation and
changes of allelic expression induced by hyperhomocysteinaemia in
patients with uraemia. Lancet. 361:1693–1699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ptak C and Petronis A: Epigenetics and
complex disease: From etiology to new therapeutics. Annu Rev
Pharmacol Toxicol. 48:257–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang Y, Chang X, Lee J, Cho YG, Zhong X,
Park IS, Liu JW, Califano JA, Ratovitski EA, Sidransky D, et al:
Cigarette smoke induces promoter methylation of single-stranded
DNA-binding protein 2 in human esophageal squamous cell carcinoma.
Int J Cancer. 128:2261–2273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakajima K, Takeoka M, Mori M, Hashimoto
S, Sakurai A, Nose H, Higuchi K, Itano N, Shiohara M, Oh T, et al:
Exercise effects on methylation of ASC gene. Int J Sports Med.
31:671–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oommen AM, Griffin JB, Sarath G and
Zempleni J: Roles for nutrients in epigenetic events. J Nutr
Biochem. 16:74–77. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duthie SJ: Folic acid deficiency and
cancer: Mechanisms of DNA instability. Br Med Bull. 55:578–592.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peterson CL and Laniel MA: Histones and
histone modifications. Curr Biol. 14:R546–R551. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jenuwein T: Re-SET-ting heterochromatin by
histone methyltransferases. Trends Cell Biol. 11:266–273. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang W, Luo XY and Sanmuels V: Gene
silencing: Double-stranded RNA mediated mRNA degradation and gene
inactivation. Cell Res. 11:181–186. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wassenegger M: The role of the RNAi
machinery in heterochromatin formation. Cell. 122:13–16. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schena FP, Serino G and Sallustio F:
MicroRNAs in kidney diseases: New promising biomarkers for
diagnosis and monitoring. Nephrol Dial Transplant. 29:755–763.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Woroniecki R, Gaikwad AB and Susztak K:
Fetal environment, epigenetics, and pediatric renal disease.
Pediatr Nephrol. 26:705–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zandi-Nejad K, Luyckx VA and Brenner BM:
Adult hypertension and kidney disease: The role of fetal
programming. Hypertension. 47:502–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luyckx VA and Brenner BM: Low birth
weight, nephron number, and kidney disease. Kidney Int Suppl.
68:S68–S77. 2005. View Article : Google Scholar
|
|
38
|
Alexander BT: Intrauterine growth
restriction and reduced glomerular number: Role of apoptosis. Am J
Physiol Regul Integr Comp Physiol. 285:R933–R934. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hughson MD, Douglas-Denton R, Bertram JF
and Hoy WE: Hypertension, glomerular number, and birth weight in
African Americans and white subjects in the southeastern United
States. Kidney Int. 69:671–678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hodgin JB, Rasoulpour M, Markowitz GS and
D'Agati VD: Very low birth weight is a risk factor for secondary
focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 4:71–76.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bechtel W, McGoohan S, Zeisberg EM, Müller
GA, Kalbacher H, Salant DJ, Müller CA, Kalluri R and Zeisberg M:
Methylation determines fibroblast activation and fibrogenesis in
the kidney. Nat Med. 16:544–550. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sayyed SG, Gaikwad AB, Lichtnekert J,
Kulkarni O, Eulberg D, Klussmann S, Tikoo K and Anders HJ:
Progressive glomerulosclerosis in type 2 diabetes is associated
with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation
and β phosphorylation at serine 10. Nephrol Dial Transplant.
25:1811–1817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gaikwad AB, Sayyed SG, Lichtnekert J,
Tikoo K and Anders HJ: Renal failure increases cardiac histone h3
acetylation, dimethylation, and phosphorylation and the induction
of cardiomyopathy-related genes in type 2 diabetes. Am J Pathol.
176:1079–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pentz ES, Lopez ML, Kim HS, Carretero O,
Smithies O and Gomez RA: Ren1d and Ren2 cooperate to preserve
homeostasis: Evidence from mice expressing GFP in place of Ren1d.
Physiol Genomics. 6:45–55. 2001.PubMed/NCBI
|
|
45
|
Gomez RA, Pentz ES, Jin X, Cordaillat M
and Lopez ML Sequeira: CBP and p300 are essential for renin cell
identity and morphological integrity of the kidney. Am J Physiol
Heart Circ Physiol. 296:H1255–H1262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shi S, Yu L, Chiu C, Sun Y, Chen J,
Khitrov G, Merkenschlager M, Holzman LB, Zhang W, Mundel P, et al:
Podocyte-selective deletion of dicer induces proteinuria and
glomerulosclerosis. J Am Soc Nephrol. 19:2159–2169. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ho JJ and Marsden PA: Dicer cuts the
kidney. J Am Soc Nephrol. 19:2043–2046. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Harvey SJ, Jarad G, Cunningham J, Goldberg
S, Schermer B, Harfe BD, McManus MT, Benzing T and Miner JH:
Podocyte-specific deletion of dicer alters cytoskeletal dynamics
and causes glomerular disease. J Am Soc Nephrol. 19:2150–2158.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ho J, Ng KH, Rosen S, Dostal A, Gregory RI
and Kreidberg JA: Podocyte-specific loss of functional microRNAs
leads to rapid glomerular and tubular injury. J Am Soc Nephrol.
19:2069–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ingrosso D, Cimmino A, Perna AF, Masella
L, De Santo NG, De Bonis ML, Vacca M, D'Esposito M, D'Urso M,
Galletti P, et al: Folate treatment and unbalanced methylation and
changes of allelic expression induced by hyperhomocysteinaemia in
patients with uraemia. Lancet. 361:1693–1699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Newman PE: Can reduced folic acid and
vitamin B12 levels cause deficient DNA methylation producing
mutations which initiate atherosclerosis? Med Hypotheses.
53:421–424. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ptak C and Petronis A: Epigenetics and
complex disease: From etiology to new therapeutics. Annu Rev
Pharmacol Toxicol. 48:257–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Griffiths EA and Gore SD: DNA
methyltransferase and histone deacetylase inhibitors in the
treatment of myelodysplastic syndromes. Semin Hematol. 45:23–30.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fukuda N: Development of gene therapies
for cardiovascular and renal diseases by nucleic acid medicines.
Med Chem. 2:13–19. 2006. View Article : Google Scholar : PubMed/NCBI
|