Introduction
Hepatocyte growth factor (HGF). HGF was cloned as a
growth factor for hepatocytes (1,2), is
identical to scatter factor (SF) and was originally discovered as a
fibroblast-derived cell motility factor for epithelial cells
(3). HGF is located at 7q21, which
spans >70 kb in length and consists of 18 exons. It encodes the
inactive pre-pro-HGF, a single chain of 728 amino acids (83 kDa),
which includes a signal sequence (1–31), a heavy
α chain (69 kDa), and a light β chain (34 kDa). The exons encode
the α chain, with four kringle structures (highly conserved triple
disulfide loop structures), a short spacer region between the α and
β chains, and the β chain (Fig. 1A)
(4–6).
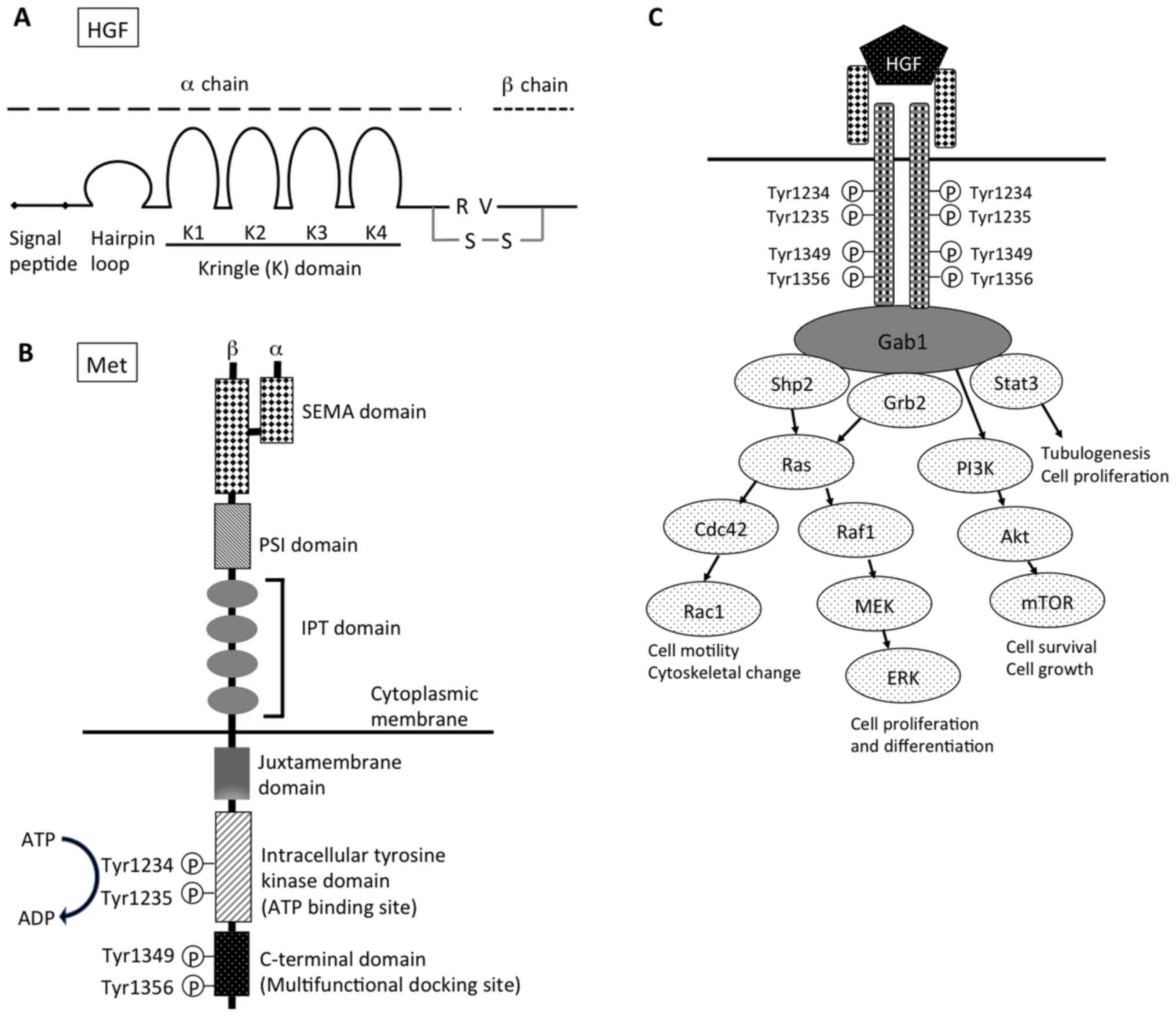 | Figure 1.Structure of (A) HGF and (B) Met. (C)
Downstream signaling pathway of HGF-Met signal. HGF, hepatocyte
growth factor; R, arginine; V, valine; S-S, disulfide bond; SEMA,
semaphorin; PSI, plexin, semaphorin, integrin cysteine-rich domain;
IPT, immunoglobulin-like regions in plexins and transcription
factors; Gab1, Grb2-associated protein 1; shp2, Src homology region
2 domain-containing phosphatase-2; Grb2, growth factor
receptor-bound protein 2; Stat3, signal transducer and activation
of transcription-3; PI3K, phosphoinositide 3-kinase; Cdc42, cell
division control protein 42 homolog; Raf1, Raf-1 proto-oncogene,
serine/threonine kinase; Rac1, Rac family small GTPase 1; MEK,
mitogen-activated protein kinase kinase; mTOR, mechanistic target
of rapamycin; ERK, extracellular signal-regulated kinases. |
HGF is produced and secreted by adjacent stromal and
mesenchymal cells, it contributes to the development of epithelial
organs in a paracrine fashion, exerts regenerative effects on
epithelia in the liver, kidney, lung, and other tissues, and
promotes the regression of fibrosis in numerous organs (7,8). Various
growth factors, cytokines, and prostaglandins upregulate HGF gene
expression, including basic fibroblast growth factor, oncostatin M,
hypoxia-inducible factor 1α and nuclear factor-κB (NF-κB) (9). By contrast, transforming growth factor
(TGF)-β1 was demonstrated to markedly downregulate HGF gene
expression (10,11).
HGF forms a family with HGF-like protein (HLP), a
unique protein with a domain structure similar to that of HGF
(12). Macrophage stimulating protein
(MSP) was discovered as a serum protein that promoted mouse
macrophage motility (13), and was
later purified to homogeneity from human plasma (14). Based on the amino acid sequence
homology and biological activity in macrophages, Shimamoto et
al (15) identified that HLP was
identical to MSP (15). MSP/HLP has a
45% amino acid sequence similarity with HGF (12) and is characterized by kringle domains
in the α chain, and a serine protease domain in the β chain;
however, it is devoid of enzymatic activity due to amino acid
substitutions in the catalytic triad.
Met
The receptor for HGF was identified as a
c-met protooncogene, which produces a transmembrane receptor
tyrosine kinase (16,17). MET is located at 7q31, which spans
>120 kb in length and consists of 21 exons (18). The MET receptor is a heterodimer,
consisting of an extracellular α-subunit (50 kDa) with the
N-terminal and β-subunit (140 kDa) that are linked by disulfide
bonds. The Met β-subunit consists of a semaphorin domain (SEMA), a
plexin, semaphorin, integrin cysteine-rich domain (PSI), four
immunoglobulin-like domains, a transmembrane region, a
juxtamembrane region, and an intracellular tyrosine kinase domain,
and a C-terminal tail (Fig. 1B)
(19,20). HGF binds the SEMA domain, in which the
MET-associated sequence resides. As a result of HGF-induced
dimerization, the intracellular tyrosine kinase domains of the two
receptor β-subunits trans-phosphorylate each other at residues
Tyr1234 and Tyr1235 within the catalytic loops (21). An intracellular multisubstrate docking
site, which is located near the C-terminal, contains tyrosine
residues, Tyr1349 and Tyr1356 (22–24). Their
subsequent phosphorylation recruits intracellular signaling
molecules, including growth factor receptor-bound protein 2 (Grb2),
Grb2-associated protein 1 (Gab1), phosphoinositide 3-kinase (PI3K),
phospholipase Cγ1, SH2 containing protein tyrosine phosphatase and
signal transducer and activation of transcription-3 (Stat3)
(Fig. 1C).
Met activation induces various biological responses,
including proliferation, motility, cell survival, morphogenesis and
angiogenesis. All of these effects are consistent with their role
in vivo. HGF and Met expression patterns indicate their
importance in the formation and homeostasis of numerous tissues. At
the early stages of development, HGF and Met exhibit expression in
the mesoderm and endoderm, respectively, and may act in an
autocrine fashion. During organogenesis, Met is detected in
epithelial cells of many organs, such as the liver, kidney, lungs
and skin.
The HGF-Met signal stimulates a wide range of
different cellular signaling pathways and it is important for the
control of tissue homeostasis under physiological conditions.
Hypoxia activates Met transcription, resulting in higher expression
levels of Met, and amplifies HGF signaling (25). However, a receptor protein-tyrosine
phosphatase (PTP), density enhanced phosphatase-1 (DEP-1), has been
implicated in the regulation of cell growth, differentiation and
transformation, and has been identified as a potential tumor
suppressor gene (26). As DEP-1
dephosphorylates particular tyrosine residues that are required for
Met-induced signaling, DEP-1 may function in controlling the
specificity of downstream signals (26). Furthermore, PTP1B knockout (KO) mice
exhibit increased insulin sensitivity and resistance to weight
gain, and are resistant to Fas-induced liver injury and lethality
(27). The downregulation of Met
involves ligand-induced internalization, ubiquitination by casitas
B-lineage lymphoma ubiquitin ligases, and lysosomal degradation
(28–30). A ubiquitination-deficient Met receptor
mutant (Y1003F) is tumorigenic and this mutant is inefficiently
targeted for degradation (29).
HGF and Met KO mice
The indispensable roles of the HGF-Met system in
mammalian development have been elucidated by the targeted
disruption of the HGF and c-met genes. HGF KO mice
were generated in previous studies (2,30). The
deletion was embryonically lethal; HGF−/− mice succumbed
between embryonic day (E)13 and E16.5, and the mice also exhibited
placental defects (31). In addition,
the c-met KO mice were embryonically lethal between E13 and
E16.5 (32). Furthermore, mutations
of two phosphorylated tyrosines (Tyr1349 and Tyr1356) in the
carboxy-terminal tail (MetD) lead to the failed coupling
of the Met receptors to their effectors. Therefore,
MetD/D mice demonstrated embryonic death with placenta,
liver and limb muscle defects (22),
mimicking the phenotype of c-met null mutants.
Conditional Met KO mice
Numerous reports of conditional KO (cKO) mice have
revealed that the HGF-Met system has developmental and regenerative
effects at various epithelial sites in the immune system, including
the liver, kidneys, lungs and neurons. Based upon these data, the
common underlying mechanisms of the HGF-Met system for tissue
repair in many organs are emphasized in the current study.
Methodology of the Cre-loxP system for
Met deletion
This selective ablation system allows gene silencing
of the c-met gene to be restricted to specific tissues and,
in certain cases, to specific times during development or
regeneration by simultaneous use of the Cre-loxP system
simultaneously (33,34). Inactivation of the mouse c-met
gene was achieved by a conditional deletion of exon 16, which
contains an ATP-binding site in the intracellular tyrosine kinase
domain, essential for activation of the HGF-Met signaling pathway
(33,34). These ‘floxed’ c-met mice were
mated with various Cre recombinase transgenic mice, and
conditional c-met KO (Met−/−; cKO) mice have been
broadly used to analyze the function of the HGF-Met signaling
pathway in development or during the regeneration process (Table I).
 | Table I.Summarized phenotypes of conditional
c-Met KO mice (Cre-LoxP system). |
Table I.
Summarized phenotypes of conditional
c-Met KO mice (Cre-LoxP system).
| Organ | Target cells | Cre-expression
(gene promoter) | Summarized
outcomes | (Refs.) |
|---|
| Liver | Hepatocytes (oval
cells) | Alb, Mx1, Alfp | Increased liver
damage and fibrosis. Impaired liver regeneration. | (33–39) |
|
|
|
| Increase of
apoptosis. Decrease of hepatocyte migration and proliferation. | (33–35) |
|
|
|
| Defects in redox
regulation. Failure of hepatic stem cell mobilization. | (36,39) |
| Kidney | Tubular cells | HoxB7 (collecting
duct), Ksp, γGT (proximal) | Reduction of uretic
bud branching and nephrons. Decreased kidney regeneration. | (40,41) |
|
|
|
| Increased
interstitial fibrosis, tubular necrosis and apoptosis. | (40,41) |
|
| Podocytes | Podocin | Increased podocyte
injury and proteinuria. | (42) |
| Lung | Alveolar type II
cells | SP-C | Impaired airspace
formation marked by reductions in alveolar epithelial cell
abundance and survival. | (50,51) |
|
|
|
| Failure of the
vascular system. | (50,51) |
| Pancreas | β-cells | RIP, Pdx | Impairment of
glucose tolerance and glucose-dependent insulin secretion. | (52,53) |
|
|
|
| Incomplete maternal
β-cell adaptation. Development of gestational diabetes
mellitus. | (79) |
|
|
|
| Sensitive to
injuries and decrease of β-cell regeneration. | (54,55) |
| Heart | Cardiomyocytes | α-MHC | Cardiomyocyte
hypertrophy associated with interstitial fibrosis and systolic
cardiac dysfunction. | (56) |
| Breast | Mammary
epithelium | MMTV | Defects in
branching in mammary glands. | (80) |
| Skin | Keratinocytes | K14 | Reepitheliazation
after skin wounding. | (57) |
| Muscle | Satellite
cells | Pax7 | Defective muscle
regeneration in response to injury. | (81) |
| Eye | Retinal
pigment | AAV injection | Reduction of
retinal pigment epithelium migration epithelium into the outer
retina of laser-injured eyes. | (82) |
| Neuron | Neurons in the
dorsal pallium | Emx1 | Alteration of
neuron architecture. Excitatory hyperconnectivity and
hypoactivity. | (45–48) |
|
| All neural
cells | Nestin | Deficit in
contextual fear condition. | (45) |
|
| Myenteric plexus
neurons | Wnt1 | Reduced length of
neurites and increased bowel injury. | (49) |
| Immunity | Dendritic
cells | Mx1 | Failure to emigrate
toward lymph nodes during inflammation. Impaired contact
hypersensitivity reaction. | (83) |
|
| Neutrophils | Mrp8 | Increased tumor
growth and metastasis. | (43) |
|
| T-cells | CD4 | Acceleration of
age-related thymic involution. | (44) |
Liver-specific Met deletion
Borowiak et al (33) reported that Cre expression was induced
by interferon-γ and complete recombination of the floxed allele
occurred in the liver of Mx-Cre;Metflox/− mice. The size
of the liver in these mice was normal, and the histology was
unchanged three weeks after recombination; however, after six
months, many small lipid vesicles were observed in the livers. More
marked hepatic changes were observed in these cKO mice during
regeneration. Following partial hepatectomy (PH), the size of the
liver in conditional Met−/− mice was smaller, and the
proliferating cell population was decreased by 60% when compared
with that of the control mice. Regarding the cell cycle, the
Met−/− livers exhibited a defective exit from quiescence
and reduced entry into the S phase. Huh et al (34) reported that the adaptive responses of
the liver to injury were markedly affected (34). These cKO mice were hypersensitive to
Fas-induced apoptosis of hepatocytes. When injected with a low dose
of anti-Fas antibody, wild-type mice survived with signs of minor
injury, whereas almost all cKO mice succumbed due to massive
apoptosis and hemorrhagic necrosis. In addition, subsequent to
injection with a single necrogenic dose of chemokine (C-C motif)
ligand 4, cKO mice exhibited impaired recovery rather than a
deficit in hepatocyte proliferation (34). The delayed regeneration was associated
with a persistent inflammatory reaction, the over-production of
osteopontin, and early and prominent dystrophic calcification
(34). These results revealed that
the disruption of c-met primarily affects hepatocyte
survival and tissue remodeling.
To address the role of HGF during the
G2/M transition, Factor et al (35) evaluated the cell cycle following PH of
cKO (Alb-Cre;Metfl/fl) mice and identified that HGF has
a novel function in the regulation of G2/M-associated gene
expression (35). These cKO mice
demonstrated defects in redox regulation, and thus the increased
sensitivity to Fas-induced apoptosis and adaptive upregulation of
NF-κB survival signaling (36). These
cKO mice were fed a methionine-choline-deficient diet to imitate
non-alcoholic-fatty liver disease, and this led to massive
steatosis, decreased survival and higher transaminase levels in
these mice (37). These findings
demonstrate that the HGF-Met signaling pathway is directly involved
in the proliferation and survival of hepatocytes.
Other functions of HGF have been reported in the
liver. The loss of the HGF-Met signaling pathway in hepatocytes
enhanced, rather than suppressed, the early stages of
hepatocarcinogenesis (38). cKO mice
treated with N-nitrosodiethylamine developed significantly more
tumors of a larger size, associated with the increased
proliferation and enhanced activation of estimated glomerular
filtration rate signaling. Focusing on the oval cells (hepatic stem
cell progeny), the liver-specific Met−/− mice were
subjected to chronic liver injury, which was induced by a diet
containing a porphyrinogenic agent (39). The absence of Met caused severe
hepatic degradation and prevented stem-cell-mediated liver
regeneration; thus, HGF may also control liver regeneration via
stem cells (39).
Kidney-specific Met deletion
In the kidneys, Met is expressed and HGF-Met
signaling is important in renal tubular epithelial cells and
visceral epithelial cells (podocytes). Ishibe et al
(40) reported the selective loss of
Met expression in the collecting system. The morphology of the
collecting system was demonstrated to be almost normal, although a
reduction in the number of nephrons and glomerular hypertrophy was
observed (40). However, to address
the role of HGF in the adult collecting ducts during renal injury,
cKO mice (HoxB7-Cre;Metfl/fl) were generated. In a model
of nephron injury and fibrosis, increased interstitial fibrosis,
inflammatory cell infiltration and acute tubular necrosis were
noted in the tubular cell-specific Met−/− mice (41). Furthermore, these mice exhibited a
reduced tubular cell proliferation and kidney regenerative capacity
following release of the obstruction, thus leading to impaired
functional recovery. Therefore, the HGF-Met signaling pathway in
the collecting duct is a major regulator of cell survival and
induction of the repair process.
Podocyte-specific Met−/− mice were also
generated in a previous study (42).
Subsequent to adriamycin treatment, these cKO mice developed more
marked podocyte injury and albuminuria.
Immune system-specific Met
deletion
The HGF-Met system has become a focus of studies on
immunity. Mutations or amplifications of Met are correlated with
the pathogenesis of various types of tumor, and Met is expressed by
cancer cells, as well as by tumor-associated stromal cells. The
HGF-Met system is also required for neutrophil chemoattraction and
cytotoxicity, and Met deletion in neutrophils enhances tumor growth
and metastasis (43). This may be due
to the reduced neutrophil infiltration of the primary tumors and
the metastatic sites. Tumor-derived TNF-α or other inflammatory
stimuli induce Met in neutrophils, and this is essential for
neutrophil transmigration across the activated endothelium and for
inducible nitric oxide synthase expression. Furthermore, nitric
oxide release by HGF-stimulated neutrophils promotes cancer cell
killing, which prevents tumor growth and metastasis.
Song et al (44) generated T cell-specific
Met−/− mice to investigate the role of the HGF-Met
signaling pathway in thymocyte development and recovery (44). These mice were more sensitive to
sub-lethal irradiation and dexamethasone treatment. The number of
total thymocytes and their subsets was markedly reduced in
T-cell-specific Met−/− mice, and the thymic architecture
of 12-month-old T-cell-specific Met−/− mice was similar
to that of the 20-month-old wild-types.
Neuron-specific Met−
deletion
The HGF-Met system regulates neuronal development,
including neuronal growth and synapse development. A5′ promoter
polymorphism of c-met is correlated with an increased risk
of autism spectrum disorder (ASD)(45), and Met expression levels are reduced
in the postmortem temporal lobe of individuals with autism and Rett
syndrome. Through human genetic analysis and murine neuroanatomical
expression mapping, HGF-Met signaling was demonstrated to be
involved in the development of forebrain circuits controlling
social and emotional behaviors that are atypical in ASD (46). To clarify roles of the HGF-Met
signaling pathway on forebrain circuit development, the cKO mice
(Emx1-Cre;Metfx/fx) with Met deletion in the dorsal
pallium-derived forebrain neurons were investigated (46). Excitatory hyperconnectivity in
specific neocortical microcircuits constitutes a basis for the
Met-mediated ASD risk (46,47). Consistent with the morphological and
biochemical changes, cKO mice exhibited the precocious maturation
of excitatory synapses, as indicated by a reduction in the
proportion of silent synapses, a faster GluN2A subunit switch, and
an enhanced acquisition of
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors at
synaptic sites (48). Mistimed
maturation of glutamatergic synapses leads to aberrant neuronal
circuits that may be associated with risk for ASD. Furthermore, to
understand the impacts of Met on behavior, Thompson and Levitt
(45) investigated two lines of cKO
(Emx1-Cre;Metfx/fx and Nestin-Cre;Metfx/fx)
mice in which Met was deleted from specific cell populations of the
central nervous system. The Emx1-Cre;Metfx/fx mice
displayed significant hypoactivity in the activity chamber and a
deficit in spontaneous alteration in the T-maze. Notably,
neuron-specific cKO mice exhibited deficits in contextual fear
conditioning (45). HGF-Met signaling
therefore contributes to the development of circuits mediating
social, emotional and cognitive behavior.
In addition, Met was localized to a subset of
calcitonin gene-associated peptide-positive myenteric plexus
neurons, which are intrinsic primary afferent neurons in the gut
(49). Met deletion in myenteric
plexus neurons demonstrated a marked loss and reduced length of
myenteric plexus Met-immunoreactive neurites (49). These mice exhibited more bowel damage
and reduced epithelial cell proliferation following dextran sodium
sulfate treatment (49).
Lung-specific Met deletion
Pulmonary capillary development depends on
epithelium-derived vascular endothelial growth factor (VEGF)-A
(50), and HGF expression may be
associated with septum formation. To investigate the essential
roles of HGF, alveolar epithelial cell type II (AECII)-specific
Met−/− mice were generated (50). The cKO (Met−/−) lung
displayed impaired saccular development and enlarged distal
airspaces with few primary septae. Furthermore, to analyze the
postnatal phenotype of these cKO mice using doxycycline,
tri-transgenic mice were generated (51). The cKO mice exhibited markedly
impaired airspace formation due to a reduction in AEC abundance and
survival, truncation of the pulmonary vascular bed and enhanced
oxidative stress in AECIIs (51). The
HGF-Met signaling pathway therefore performs essential functions in
lung development, particularly in septum formation.
Pancreatic β-cell-specific Met
deletion
To examine the essential roles of the HGF-Met system
in β-cells, Met was deleted using rat insulin II promoter
(RIP)-driven Cre expression (52,53).
β-cell-specific Met−/− mice exhibited normal body
weight, blood glucose and plasma insulin levels (52,53).
However, the mice exhibited reduced glucose tolerance and reduced
plasma insulin levels following glucose challenge; thus, the
HGF-Met signaling pathway may be essential for normal
glucose-dependent insulin secretion. In addition, cKO mice
displayed markedly increased apoptosis and decreased proliferation
following multiple low-dose streptozotocin treatments, and markedly
reduced β-cell regeneration following pancreatectomy (54,55).
Cardiac myocyte-specific Met
deletion
To investigate the requirement of the HGF-Met
signaling pathway in cardiomyocytes, Arechederra et al
(56) generated mice with Met
deletion in cardiomyocytes using the α-myosin heavy chain (MHC)-Cre
mouse line. These cKO mice developed cardiomyocyte hypertrophy and
interstitial fibrosis. The mice displayed significant upregulation
of markers of myocardial damage, such as α-MHC and atrial
natriuretic factor, and systolic cardiac dysfunction.
Skin-specific Met deletion
The skin has barrier functions for many forms of
environmental stress, and therefore has an efficient system to
repair wounds. At the wound edges, keratinocytes form a
hyperproliferative epithelium (HE) that strongly proliferates and
migrates to recover the wound area, and HGF and Met are upregulated
in the HE during wound repair. Chmielowiec et al (57) analyzed the epidermis in cKO
(K14-Cre;Metfl/fl) mice (57). The reepithelialization of skin
keratinocytes was impaired, wound closure was slightly attenuated
in keratinocyte-specific Met−/− mice and the closure of
a scratch wound occurred in the presence of only a few remaining
Met-positive cells (57). Therefore,
the HGF-Met signaling pathway is a fundamental regenerative process
in the skin.
Specific inhibition of HGF-Met
downstream signaling
Among the downstream signaling molecules, Gab1 is
critical in HGF-dependent biological responses (58,59). Gab1
is a scaffolding adaptor protein, and a direct and robust
interaction of Gab1 and Met is responsible for the various
biological activities of HGF. The phosphorylation of C-terminal
tyrosine residues in the docking site of Met recruits intracellular
signaling molecules, including PI3K, Src, Grb2, Shc adaptor, and
the multi-adaptor Gab1. To address the role of specific signaling
pathways in the HGF-Met system in vivo, these
multifunctional docking sites were converted into specific binding
motifs for PI3K, Src or Grb2 (Met2P, Met2S or
Met2G) (60). All three
Met mutants retained normal signaling, but recruited specific
effectors differentially. Met2G mice developed normally,
but Met2P and Met2S mice displayed different
phenotypes and rescued of distinct tissues following
loss-of-function (60). The partial
rescue of myoblast migration was the only trait in common and it
most likely resulted from the net contribution of residual
Gab1-mediated Met signaling.
HGF transgenic mice
Liver-specific HGF overexpression
Transgenic mice with HGF overexpression (HGF-Tg)
under the control of the metallothionein promoter exhibited an
increased liver size and a marked increase of 2N small hepatocytes
(61). HGF-Tg mice made using the
albumin promoter exhibit a lower expression level of HGF when
compared with HGF-Tg mice made using the metallothionein promoter,
with a smaller increase in liver size (62,63). The
overexpression of HGF exerts a protective effect against
Fas-mediated hepatic apoptosis in HGF-Tg mice. Akt phosphorylation
and B-cell lymphoma-extra large expression levels were increased in
HGF-Tg mice before and after anti-Fas antibody injection (64). Activation of the microsomal
triglyceride transfer protein and apolipoprotein B, accompanied by
higher triglyceride levels in the serum were also observed in HGF
transgenic mice (65).
Pancreas-specific HGF
overexpression
HGF-Tg mice were used to investigate HGF functions
in the pancreas. The islets of RIP-regulated HGF-Tg mice contain
more insulin per β-cell, secrete more insulin in response to
glucose, have higher steady-state glucose transporter 2 and
glucokinase levels, and take up and metabolize glucose more
effectively. Furthermore, HGF has positive effects on β-cell
mitogenesis, glucose sensing, β-cell markers of differentiation and
transplant survival (66). Insulin
receptor substrate-2 (IRS2)−/− mice develop diabetes due
to insulin resistance and β-cell failure. After crossing HGF-Tg
mice with IRS2 KO mice, the progeny exhibited significantly reduced
hyperglycemia, compensatory hyperinsulinemia, improved glucose
tolerance and increased glucose-stimulated insulin secretion in
Tg/KO islets. Additionally, β-cell proliferation and mass were
increased, and the mortality rate was decreased (67,68).
HGF-Tg mice were relatively hypoglycemic in post-prandial and
fasting states, and demonstrated a markedly attenuated response to
the diabetogenic effects of streptozotocin (68).
Neuron-specific HGF
overexpression
Amyotrophic lateral sclerosis (ALS) is an
adult-onset neurodegenerative disease characterized by a
progressive loss of motoneurons in the brain cortex and spinal
cord. HGF overexpression in the nervous system attenuates
motoneuron death and axonal degeneration. HGF overexpressing mice
exhibit an extended life span in an ALS model (G93A mice) that
overexpresses mutated Cu2+/Zn2+ superoxide
dismutase. HGF alleviates the symptoms of ALS by direct
neurotrophic effects on motoneurons and indirect effects on glial
cells, possibly favoring a reduction in glutamatergic neurotoxicity
(69). G93A/HGF-Tg mice demonstrated
marked decreases in the numbers of microglia and reactive
astrocytes, and an attenuation of the motoneuron loss. HGF
overexpression prevented monocyte chemoattractant protein-1
induction, suppressed caspase activation, and increased expression
of X chromosome-linked inhibition of apoptosis protein in the
motoneurons of G93A mice (70).
Spinal and bulbar muscular atrophy (SBMA) is an
inherited motor neuron disease in the brain stem and spinal cord,
caused by the expansion of a polyglutamine tract in the androgen
receptor (AR) protein, which diffusely accumulates as inclusions in
nuclei. By crossing SBMA model mice expressing a mutated AR gene
with HGF-Tg mice, Ding et al (71) demonstrated that the high level of HGF
expression induced Akt phosphorylation and modestly ameliorated
motor symptoms in an SBMA mouse model. HGF administration may
provide a possible combination therapy with other disease-modifying
therapeutic strategies in SBMA (71).
Notably, HGF overexpression in the central nervous
system advances learning and memory performance (72). HGF upregulated N-methyl-D-aspartate
receptor subunits, NR2A and NR2B, and normal nervous system
plasticity (72).
Mammary gland-specific HGF
overexpression
The expressions levels of HGF and Met are regulated
during mouse mammary gland development. HGF-Tg mice exhibited a
range of alterations in the architecture of virgin mouse mammary
glands, including an enhancement of the ductal end bud size, and
numbers and hyperplastic branching morphogenesis (73).
Kidney-, skin-, and muscle-specific
HGF overexpression
Concerning the role of HGF in the kidneys, HGF-Tg
mice with expression in the proximal tubules, under the direction
of the γ-glutamyl transpeptidase-I promoter, were developed. HGF
overexpression markedly protected kidneys from ischemia-induced
acute renal failure (74).
By analyzing excisional wound sites in HGF-Tg mice
under the metallothionein promoter, Toyoda et al (75) revealed that HGF enhanced granulation
tissue accompanied by marked vascularization with the induction of
VEGF (75).
Muscle-specific HGF-Tg (SK-HGF) mice did not exhibit
altered plasma HGF levels. When HGF-Tg mice were fed a normal diet,
these mice displayed similar levels in body weight and blood
glucose, plasma triglycerides and plasma insulin levels, and
glucose tolerance when compared with wild-types. Obese HGF-Tg mice
recovered improved whole-body glucose tolerance. Thus,
muscle-specific expression of HGF counteracts obesity-mediated
muscle insulin resistance and glucose tolerance in mice (76).
Conclusion
Numerous studies have broadly examined biological
functions of the HGF-Met signaling pathway. Various studies have
demonstrated the crucial physiological roles and therapeutic
potentials of HGF during fetal development and recovery from
disease conditions, including acute and chronic tissue injury, and
immunological and neurodegenerative diseases. The characterization
of mice with cell/tissue-selective disruption of the c-met
gene particularly elucidated the essential roles of the HGF-Met
signaling pathway in proliferation, survival, morphogenesis, tissue
development, regeneration and organ homeostasis. Furthermore, the
examination of mice with organ-specific overexpression of HGF
revealed the therapeutic potential of using HGF to treat various
types of disease. It is hoped that this review leads to important
discussion of HGF therapeutic strategies based upon scientific
evidence.
Acknowledgements
The author would like to thank Dr Matsumoto for
useful advice.
Glossary
Abbreviations
Abbreviations:
|
AEC
|
alveolar epithelial cells
|
|
ALS
|
amyotrophic lateral sclerosis
|
|
AR
|
androgen receptor
|
|
ASD
|
autism spectrum disorder
|
|
cKO
|
conditional knockout
|
|
Gab1
|
Grb2-associated protein 1
|
|
Grb2
|
growth factor receptor-bound protein
2
|
|
HE
|
hyperproliferative epithelium
|
|
HGF
|
hepatocyte growth factor
|
|
HLP
|
HGF-like protein
|
|
IPT
|
immunoglobulin-like regions in plexins
and transcription factors
|
|
MSP
|
macrophage stimulating protein
|
|
PH
|
partial hepatectomy
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
PSI
|
plexin, semaphorin, integrin
cysteine-rich domain
|
|
RIP
|
rat insulin II promoter
|
|
SBMA
|
spinal and bulbar muscular atrophy
|
|
SEMA
|
semaphorin
|
|
SF
|
scatter factor
|
|
Shp2
|
Src homology region 2
domain-containing phosphatase-2
|
|
Stat3
|
signal transducer and activation of
transcription-3
|
|
Tg
|
transgenic
|
References
|
1
|
Nakamura T, Nishizawa T, Hagiya M, Seki T,
Shimonishi M, Sugimura A, Tashiro K and Shimizu S: Molecular
cloning and expression of human hepatocyte growth factor. Nature.
342:440–443. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyazawa K, Tsubouchi H, Naka D, Takahashi
K, Okigaki M, Arakaki N, Nakayama H, Hirono S, Sakiyama O,
Takahashi K, et al: Molecular cloning and sequence analysis of cDNA
for human hepatocyte growth factor. Biochem Biophys Res Commun.
163:967–973. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stoker M, Gherardi E, Perryman M and Gray
J: Scatter factor is a fibroblast-derived modulator of epithelial
cell mobility. Nature. 327:239–242. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zarnegar R, Petersen B, DeFrances MC and
Michalopoulos G: Localization of hepatocyte growth factor (HGF)
gene on human chromosome 7. Genomics. 12:147–150. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fukuyama R, Ichijoh Y, Minoshima S,
Kitamura N and Shimizu N: Regional localization of the hepatocyte
growth factor (HGF) gene to human chromosome 7 band q21.1.
Genomics. 11:410–415. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seki T, Hagiya M, Shimonishi M, Nakamura T
and Shimizu S: Organization of the human hepatocyte growth
factor-encoding gene. Gene. 102:213–219. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Andermarcher E, Surani MA and Gherardi E:
Co-expression of the HGF/SF and c-met genes during early mouse
embryogenesis precedes reciprocal expression in adjacent tissues
during organogenesis. Dev Genet. 18:254–266. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matsumoto K, Funakoshi H, Takahashi H and
Sakai K: HGF-Met pathway in regeneration and drug discovery.
Biomedicines. 2:275–300. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fajardo-Puerta AB, Mato Prado M, Frampton
AE and Jiao LR: Gene of the month: HGF. J Clin Pathol. 69:575–579.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsumoto K, Tajima H, Okazaki H and
Nakamura T: Negative regulation of hepatocyte growth factor gene
expression in human lung fibroblasts and leukemic cells by
transforming growth factor-beta 1 and glucocorticoids. J Biol Chem.
267:24917–24920. 1992.PubMed/NCBI
|
|
11
|
Harrison P, Bradley L and Bomford A:
Mechanism of regulation of HGF/SF gene expression in fibroblasts by
TGF-beta1. Biochem Biophys Res Commun. 271:203–211. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han S, Stuart LA and Degen SJ:
Characterization of the DNF15S2 locus on human chromosome 3:
Identification of a gene coding for four kringle domains with
homology to hepatocyte growth factor. Biochemistry. 30:9768–9780.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leonard EJ and Skeel A: A serum protein
that stimulates macrophage movement, chemotaxis and spreading. Exp
Cell Res. 102:434–438. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Skeel A, Yoshimura T, Showalter SD, Tanaka
S, Appella E and Leonard EJ: Macrophage stimulating protein:
Purification, partial amino acid sequence, and cellular activity. J
Exp Med. 173:1227–1234. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimamoto A, Kimura T, Matsumoto K and
Nakamura T: Hepatocyte growth factor-like protein is identical to
macrophage stimulating protein. FEBS Lett. 333:61–66. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bottaro DP, Rubin JS, Faletto DL, Chan AM,
Kmiecik TE, Vande Woude GF and Aaronson SA: Identification of the
hepatocyte growth factor receptor as the c-met proto-oncogene
product. Science. 251:802–804. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Naldini L, Vigna E, Narsimhan RP, Gaudino
G, Zarnegar R, Michalopoulos GK and Comoglio PM: Hepatocyte growth
factor (HGF) stimulates the tyrosine kinase activity of the
receptor encoded by the proto-oncogene c-MET. Oncogene. 6:501–504.
1991.PubMed/NCBI
|
|
18
|
Liu Y: The human hepatocyte growth factor
receptor gene: Complete structural organization and promoter
characterization. Gene. 215:159–169. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Niemann HH: Structural insights into Met
receptor activation. Eur J Cell Biol. 90:972–981. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stamos J, Lazarus RA, Yao X, Kirchhofer D
and Wiesmann C: Crystal structure of the HGF beta-chain in complex
with the Sema domain of the Met receptor. EMBO J. 23:2325–2335.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rodrigues GA and Park M:
Autophosphorylation modulates the kinase activity and oncogenic
potential of the Met receptor tyrosine kinase. Oncogene.
9:2019–2027. 1994.PubMed/NCBI
|
|
22
|
Maina F, Casagranda F, Audero E, Simeone
A, Comoglio PM, Klein R and Ponzetto C: Uncoupling of Grb2 from the
Met receptor in vivo reveals complex roles in muscle development.
Cell. 87:531–542. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ponzetto C, Bardelli A, Zhen Z, Maina F,
dalla Zonca P, Giordano S, Graziani A, Panayotou G and Comoglio PM:
A multifunctional docking site mediates signaling and
transformation by the hepatocyte growth factor/scatter factor
receptor family. Cell. 77:261–271. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sachs M, Weidner KM, Brinkmann V, Walther
I, Obermeier A, Ullrich A and Birchmeier W: Motogenic and
morphogenic activity of epithelial receptor tyrosine kinases. J
Cell Biol. 133:1095–1107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palka HL, Park M and Tonks NK: Hepatocyte
growth factor receptor tyrosine kinase met is a substrate of the
receptor protein-tyrosine phosphatase DEP-1. J Biol Chem.
278:5728–5735. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sangwan V, Paliouras GN, Cheng A, Dubé N,
Tremblay ML and Park M: Protein-tyrosine phosphatase 1B deficiency
protects against Fas-induced hepatic failure. J Biol Chem.
281:221–228. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peschard P and Park M: Escape from
Cbl-mediated downregulation: A recurrent theme for oncogenic
deregulation of receptor tyrosine kinases. Cancer Cell. 3:519–523.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Abella JV, Peschard P, Naujokas MA, Lin T,
Saucier C, Urbé S and Park M: Met/hepatocyte growth factor receptor
ubiquitination suppresses transformation and is required for Hrs
phosphorylation. Mol Cell Biol. 25:9632–9645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmidt C, Bladt F, Goedecke S, Brinkmann
V, Zschiesche W, Sharpe M, Gherardi E and Birchmeier C: Scatter
factor/hepatocyte growth factor is essential for liver development.
Nature. 373:699–702. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Uehara Y, Minowa O, Mori C, Shiota K, Kuno
J, Noda T and Kitamura N: Placental defect and embryonic lethality
in mice lacking hepatocyte growth factor/scatter factor. Nature.
373:702–705. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bladt F, Riethmacher D, Isenmann S, Aguzzi
A and Birchmeier C: Essential role for the c-met receptor in the
migration of myogenic precursor cells into the limb bud. Nature.
376:768–771. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Borowiak M, Garratt AN, Wüstefeld T,
Strehle M, Trautwein C and Birchmeier C: Met provides essential
signals for liver regeneration. Proc Natl Acad Sci USA. 101:pp.
10608–10613. 2004, View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huh CG, Factor VM, Sánchez A, Uchida K,
Conner EA and Thorgeirsson SS: Hepatocyte growth factor/c-met
signaling pathway is required for efficient liver regeneration and
repair. Proc Natl Acad Sci USA. 101:pp. 4477–4482. 2004, View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Factor VM, Seo D, Ishikawa T, Kaposi-Novak
P, Marquardt JU, Andersen JB, Conner EA and Thorgeirsson SS: Loss
of c-Met disrupts gene expression program required for G2/M
progression during liver regeneration in mice. PLoS One.
5:e127392010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gómez-Quiroz LE, Factor VM, Kaposi-Novak
P, Coulouarn C, Conner EA and Thorgeirsson SS: Hepatocyte-specific
c-Met deletion disrupts redox homeostasis and sensitizes to
Fas-mediated apoptosis. J Biol Chem. 283:14581–14589. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kroy DC, Schumacher F, Ramadori P, Hatting
M, Bergheim I, Gassler N, Boekschoten MV, Müller M, Streetz KL and
Trautwein C: Hepatocyte specific deletion of c-Met leads to the
development of severe non-alcoholic steatohepatitis in mice. J
Hepatol. 61:883–890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Takami T, Kaposi-Novak P, Uchida K,
Gomez-Quiroz LE, Conner EA, Factor VM and Thorgeirsson SS: Loss of
hepatocyte growth factor/c-Met signaling pathway accelerates early
stages of N-nitrosodiethylamine induced hepatocarcinogenesis.
Cancer Res. 67:9844–9851. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ishikawa T, Factor VM, Marquardt JU, Raggi
C, Seo D, Kitade M, Conner EA and Thorgeirsson SS: Hepatocyte
growth factor/c-met signaling is required for stem-cell-mediated
liver regeneration in mice. Hepatology. 55:1215–1226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ishibe S, Karihaloo A, Ma H, Zhang J,
Marlier A, Mitobe M, Togawa A, Schmitt R, Czyczk J, Kashgarian M,
et al: Met and the epidermal growth factor receptor act
cooperatively to regulate final nephron number and maintain
collecting duct morphology. Development. 136:337–345. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma H, Saenko M, Opuko A, Togawa A, Soda K,
Marlier A, Moeckel GW, Cantley LG and Ishibe S: Deletion of the Met
receptor in the collecting duct decreases renal repair following
ureteral obstruction. Kidney Int. 76:868–876. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dai C, Saleem MA, Holzman LB, Mathieson P
and Liu Y: Hepatocyte growth factor signaling ameliorates podocyte
injury and proteinuria. Kidney Int. 77:962–973. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Finisguerra V, Di Conza G, Di Matteo M,
Serneels J, Costa S, Thompson AA, Wauters E, Walmsley S, Prenen H,
Granot Z, et al: MET is required for the recruitment of
anti-tumoural neutrophils. Nature. 522:349–353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song Y, Su M, Panchatsharam P, Rood D and
Lai L: c-Met signalling is required for efficient postnatal thymic
regeneration and repair. Immunology. 144:245–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thompson BL and Levitt P: Complete or
partial reduction of the Met receptor tyrosine kinase in distinct
circuits differentially impacts mouse behavior. J Neurodev Disord.
7:352015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Judson MC, Eagleson KL, Wang L and Levitt
P: Evidence of cell-nonautonomous changes in dendrite and dendritic
spine morphology in the met-signaling-deficient mouse forebrain. J
Comp Neurol. 518:4463–4478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qiu S, Anderson CT, Levitt P and Shepherd
GM: Circuit-specific intracortical hyperconnectivity in mice with
deletion of the autism-associated Met receptor tyrosine kinase. J
Neurosci. 31:5855–5864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qiu S, Lu Z and Levitt P: MET receptor
tyrosine kinase controls dendritic complexity, spine morphogenesis,
and glutamatergic synapse maturation in the hippocampus. J
Neurosci. 34:16166–16179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Avetisyan M, Wang H, Schill EM, Bery S,
Grider JR, Hassell JA, Stappenbeck T and Heuckeroth RO: Hepatocyte
growth factor and met support mouse enteric nervous system
development, the peristaltic response, and intestinal epithelial
proliferation in response to injury. J Neurosci. 35:11543–11558.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamamoto H, Yun EJ, Gerber HP, Ferrara N,
Whitsett JA and Vu TH: Epithelial-vascular cross talk mediated by
VEGF-A and HGF signaling directs primary septae formation during
distal lung morphogenesis. Dev Biol. 308:44–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Calvi C, Podowski M, Lopez-Mercado A,
Metzger S, Misono K, Malinina A, Dikeman D, Poonyagariyon H,
Ynalvez L, Derakhshandeh R, et al: Hepatocyte growth factor, a
determinant of airspace homeostasis in the murine lung. PLoS Genet.
9:e10032282013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Roccisana J, Reddy V, Vasavada RC,
Gonzalez-Pertusa JA, Magnuson MA and Garcia-Ocaña A: Targeted
inactivation of hepatocyte growth factor receptor c-met in
beta-cells leads to defective insulin secretion and GLUT-2
downregulation without alteration of beta-cell mass. Diabetes.
54:2090–2102. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dai C, Huh CG, Thorgeirsson SS and Liu Y:
Beta-cell-specific ablation of the hepatocyte growth factor
receptor results in reduced islet size, impaired insulin secretion,
and glucose intolerance. Am J Pathol. 167:429–436. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mellado-Gil J, Rosa TC, Demirci C,
Gonzalez-Pertusa JA, Velazquez-Garcia S, Ernst S, Valle S, Vasavada
RC, Stewart AF, Alonso LC and Garcia-Ocaña A: Disruption of
hepatocyte growth factor/c-Met signaling enhances pancreatic
beta-cell death and accelerates the onset of diabetes. Diabetes.
60:525–536. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Alvarez-Perez JC, Ernst S, Demirci C,
Casinelli GP, Mellado-Gil JM, Rausell-Palamos F, Vasavada RC and
Garcia-Ocaña A: Hepatocyte growth factor/c-Met signaling is
required for β-cell regeneration. Diabetes. 63:216–223. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Arechederra M, Carmona R, González-Nuñez
M, Gutiérrez-Uzquiza A, Bragado P, Cruz-González I, Cano E,
Guerrero C, Sánchez A, López-Novoa JM, et al: Met signaling in
cardiomyocytes is required for normal cardiac function in adult
mice. Biochim Biophys Acta. 1832:2204–2215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chmielowiec J, Borowiak M, Morkel M,
Stradal T, Munz B, Werner S, Wehland J, Birchmeier C and Birchmeier
W: c-Met is essential for wound healing in the skin. J Cell Biol.
177:151–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Weidner KM, Di Cesare S, Sachs M,
Brinkmann V, Behrens J and Birchmeier W: Interaction between Gab1
and the c-Met receptor tyrosine kinase is responsible for
epithelial morphogenesis. Nature. 384:173–176. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sachs M, Brohmann H, Zechner D, Müller T,
Hülsken J, Walther I, Schaeper U, Birchmeier C and Birchmeier W:
Essential role of Gab1 for signaling by the c-Met receptor in vivo.
J Cell Biol. 150:1375–1384. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Maina F, Panté G, Helmbacher F, Andres R,
Porthin A, Davies AM, Ponzetto C and Klein R: Coupling Met to
specific pathways results in distinct developmental outcomes. Mol
Cell. 7:1293–1306. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sakata H, Takayama H, Sharp R, Rubin JS,
Merlino G and LaRochelle WJ: Hepatocyte growth factor/scatter
factor overexpression induces growth, abnormal development, and
tumor formation in transgenic mouse livers. Cell Growth Differ.
7:1513–1523. 1996.PubMed/NCBI
|
|
62
|
Shiota G, Wang TC, Nakamura T and Schmidt
EV: Hepatocyte growth factor in transgenic mice: Effects on
hepatocyte growth, liver regeneration and gene expression.
Hepatology. 19:962–972. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Di Renzo MF, Olivero M, Martone T, Maffe
A, Maggiora P, Stefani AD, Valente G, Giordano S, Cortesina G and
Comoglio PM: Somatic mutations of the MET oncogene are selected
during metastatic spread of human HNSC carcinomas. Oncogene.
19:1547–1555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Suzuki H, Toyoda M, Horiguchi N, Kakizaki
S, Ohyama T, Takizawa D, Ichikawa T, Sato K, Takagi H and Mori M:
Hepatocyte growth factor protects against Fas-mediated liver
apoptosis in transgenic mice. Liver Int. 29:1562–1568. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kosone T, Takagi H, Horiguchi N, Ariyama
Y, Otsuka T, Sohara N, Kakizaki S, Sato K and Mori M: HGF
ameliorates a high-fat diet-induced fatty liver. Am J Physiol
Gastrointest Liver Physiol. 293:G204–G210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
García-Ocaña A, Vasavada RC, Cebrian A,
Reddy V, Takane KK, López-Talavera JC and Stewart AF: Transgenic
overexpression of hepatocyte growth factor in the beta-cell
markedly improves islet function and islet transplant outcomes in
mice. Diabetes. 50:2752–2762. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Alvarez-Perez JC, Rosa TC, Casinelli GP,
Valle SR, Lakshmipathi J, Rosselot C, Rausell-Palamos F, Vasavada
RC and García-Ocaña A: Hepatocyte growth factor ameliorates
hyperglycemia and corrects β-cell mass in IRS2-deficient mice. Mol
Endocrinol. 28:2038–2048. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Garcia-Ocaña A, Takane KK, Syed MA,
Philbrick WM, Vasavada RC and Stewart AF: Hepatocyte growth factor
overexpression in the islet of transgenic mice increases beta cell
proliferation, enhances islet mass, and induces mild hypoglycemia.
J Biol Chem. 275:1226–1232. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sun W, Funakoshi H and Nakamura T:
Overexpression of HGF retards disease progression and prolongs life
span in a transgenic mouse model of ALS. J Neurosci. 22:6537–6548.
2002.PubMed/NCBI
|
|
70
|
Kadoyama K, Funakoshi H, Ohya W and
Nakamura T: Hepatocyte growth factor (HGF) attenuates gliosis and
motoneuronal degeneration in the brainstem motor nuclei of a
transgenic mouse model of ALS. Neurosci Res. 59:446–456. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ding Y, Adachi H, Katsuno M, Huang Z,
Jiang YM, Kondo N, Iida M, Tohnai G, Nakatsuji H, Funakoshi H, et
al: Overexpression of hepatocyte growth factor in SBMA model mice
has an additive effect on combination therapy with castration.
Biochem Biophys Res Commun. 468:677–683. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kato T, Funakoshi H, Kadoyama K, Noma S,
Kanai M, Ohya-Shimada W, Mizuno S, Doe N, Taniguchi T and Nakamura
T: Hepatocyte growth factor overexpression in the nervous system
enhances learning and memory performance in mice. J Neurosci Res.
90:1743–1755. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yant J, Buluwela L, Niranjan B, Gusterson
B and Kamalati T: In vivo effects of hepatocyte growth
factor/scatter factor on mouse mammary gland development. Exp Cell
Res. 241:476–481. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fiaschi-Taesch NM, Santos S, Reddy V, Van
Why SK, Philbrick WF, Ortega A, Esbrit P, Orloff JJ and
Garcia-Ocaña A: Prevention of acute ischemic renal failure by
targeted delivery of growth factors to the proximal tubule in
transgenic mice: The efficacy of parathyroid hormone-related
protein and hepatocyte growth factor. J Am Soc Nephrol. 15:112–125.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Toyoda M, Takayama H, Horiguchi N, Otsuka
T, Fukusato T, Merlino G, Takagi H and Mori M: Overexpression of
hepatocyte growth factor/scatter factor promotes vascularization
and granulation tissue formation in vivo. FEBS Lett. 509:95–100.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sanchez-Encinales V, Cozar-Castellano I,
Garcia-Ocaña A and Perdomo G: Targeted delivery of HGF to the
skeletal muscle improves glucose homeostasis in diet-induced obese
mice. J Physiol Biochem. 71:795–805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhou D, Tan RJ, Lin L, Zhou L and Liu Y:
Activation of hepatocyte growth factor receptor, c-met, in renal
tubules is required for renoprotection after acute kidney injury.
Kidney Int. 84:509–520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mason S, Hader C, Marlier A, Moeckel G and
Cantley LG: Met activation is required for early cytoprotection
after ischemic kidney injury. J Am Soc Nephrol. 25:329–337. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Demirci C, Ernst S, Alvarez-Perez JC, Rosa
T, Valle S, Shridhar V, Casinelli GP, Alonso LC, Vasavada RC and
García-Ocana A: Loss of HGF/c-Met signaling in pancreatic β-cells
leads to incomplete maternal β-cell adaptation and gestational
diabetes mellitus. Diabetes. 61:1143–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Garner OB, Bush KT, Nigam KB, Yamaguchi Y,
Xu D, Esko JD and Nigam SK: Stage-dependent regulation of mammary
ductal branching by heparan sulfate and HGF-cMet signaling. Dev
Biol. 355:394–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Webster MT and Fan CM: c-MET regulates
myoblast motility and myocyte fusion during adult skeletal muscle
regeneration. PLoS One. 8:e817572013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kasaoka M, Ma J and Lashkari K: c-Met
modulates RPE migratory response to laser-induced retinal injury.
PLoS One. 7:e407712012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baek JH, Birchmeier C, Zenke M and
Hieronymus T: The HGF receptor/Met tyrosine kinase is a key
regulator of dendritic cell migration in skin immunity. J Immunol.
189:1699–1707. 2012. View Article : Google Scholar : PubMed/NCBI
|














