Introduction
According to data from the International Diabetes
Federation, 382 million individuals presented with diabetes
mellitus in 2013 and this number is expected to rise to 592 million
by 2035 (1). As a progressive
metabolic disorder, chronic complications occur in the late stage
of diabetes, including atherosclerosis, diabetic neuropathy,
diabetic nephropathy, diabetic retinopathy and diabetic
cardiomyopathy (DCM) (2). Among the
vast array of long-term complications associated with diabetes,
cardiovascular diseases account for the major cause of morbidity
and mortality among the diabetic population worldwide (3–7).
The Framingham Heart Study demonstrated that
diabetes was an independent risk factor of cardiovascular disease,
including heart failure (8). The
primary causes of diabetes-associated heart failure were believed
to be coronary atherosclerosis and ischemia. However, in 1972,
Rubler et al (9) reported
autopsy data of four diabetic patients with heart failure, and no
specific cause, such as coronary disease, hypertension, alcohol
consumption, or other structural heart disease was identified. The
authors subsequently introduced the term DCM. DCM is a newly
identified disease and is considered to be completely different
from coronary heart disease. Coronary heart disease usually results
from atherosclerosis of the coronary artery and it is regarded as a
type of macrovascular complication of diabetes. Diabetes promotes
the onset and development of coronary heart disease together with
numerous other factors, such as obesity, smoking and
hyperlipidemia. However, the distinct features of DCM are cardiac
hypertrophy and myocardial fibrosis in the absence of obvious
pathogenic coronary abnormalities. It appears asymptomatic until
the very late stage and left ventricular diastolic dysfunction with
preserved systolic function is an early sign of DCM, while systolic
dysfunction eventually occurs.
The pathogenesis of DCM is complex and has not been
well understood until recently. Impaired calcium handling, altered
metabolism, increased oxidative stress, remodeling of extracellular
matrix (ECM), endothelial dysfunction and mitochondrial dysfunction
have been found to participate in the pathogenesis of DCM (2,10–14). A number of signaling proteins and
pathways have been implicated in contributing to the development of
DCM, including protein kinase C, nuclear factor-κB, peroxisome
proliferator-activated receptor α, phosphatidylinositol 3-kinase
(PI3K) and mitogen activated protein kinase (MAPK) signaling
pathways (15,16).
In this context, microRNAs (miRs or miRNAs) are
found to be important in the pathogenesis of DCM. miRs were
initially described by Lee et al (17) in nematodes, Caenorhabditis elegans in
1993. As a novel family of highly conserved, short (~18–25
nucleotides), non-coding, single-stranded RNA molecules, miRs
regulate transcriptional and post-transcriptional gene expression
through binding to the 3′-untranslated region (3′-UTR) of their
target mRNA (3). Given that miRs are
crucially involved in numerous critical biological processes,
including cell proliferation, apoptosis, necrosis, migration and
differentiation, dysregulated miRs contribute to various human
diseases, including diabetes and cardiovascular diseases (4,18–20). miR-126, miR-17, miR-92a, miR-145,
miR-155, miR-133 and miR-208a were identified to be associated with
coronary artery disease; miR-1, miR-21, miR-208, miR-133a/b and
miR-499 were identified as important in the pathogenesis of acute
cardiac infarction. In addition, miRs associated with heart failure
include miR-24, miR-125b, miR-195, miR-199a and miR-214 (20–22).
Recent studies have demonstrated an association
between miRs and DCM (23–25). The expression level of miR in the
hearts of patients with DCM was found to be different when compared
with that of healthy individuals (25,26).
Furthermore, analysis of miR expression levels in the hearts of
various rat and mouse diabetic models also indicated the abnormal
expression of miRNA. Further studies demonstrated that miRs
contribute to numerous important pathophysiological processes of
DCM, including cardiomyocyte hypertrophy, myocardial fibrosis,
cardiomyocyte apoptosis, mitochondrial dysfunction, myocardial
electrical remodeling and epigenetic modification (27–32). The
present review discusses the possible role of miRs in the
pathogenesis of DCM regarding the above-mentioned processes.
miR involvement in the pathogenesis of
DCM
miRs in cardiomyocyte hypertrophy
Cardiomyocyte hypertrophy is one of the distinct
structural features of DCM. Studies have shown that various miRs
were dysregulated and contributed to the pathogenesis of
cardiomyocyte hypertrophy in DCM (29,33–36). miR-30c, miR-133a, miR-150 and miR-373
were found to be downregulated in the heart of DCM, while miR-451
was found to be upregulated (29,33–36).
miR-133 is abundantly expressed in heart tissue, and
is known to regulate various physiological and pathophysiological
events, including non-diabetic cardiac hypertrophy (37–39).
Hyperglycemia results in cardiac hypertrophy. A recent study
reported that the expression level of miR-133a was reduced in
cardiomyocytes treated with high levels of glucose, as well as in
hypertrophic cardiac tissue samples from streptozotocin
(STZ)-induced diabetic mice, and transfection of miR-133a mimics
prevented altered gene expression and hypertrophic changes
(33). Therefore, it was concluded
that miR-133a participated in mediating glucose-induced
cardiomyocyte hypertrophy in diabetes. Additionally, another study
demonstrated that serum and glucocorticoid-regulated kinase 1 and
insulin-like growth factor-1 (IGF-1) receptor may be involved in
this process as potential targets of miR-133a (33).
Various anti- and pro-growth signaling pathways have
been demonstrated to participate in cardiomyocyte hypertrophy,
including the PI3K/AKT signaling pathway. p21-activated kinases
(PAKs) and cell division control protein 42 homolog (Cdc42) are
components of the PI3K/AKT signaling pathway, and PAKs are
effectors of Cdc42. Myocardial Cdc42 and Pak1 mRNA
and protein expression levels were found to be significantly
increased in DCM rats with cardiac hypertrophy and in high glucose
(HG)-treated cardiomyocytes, which was accompanied by a significant
decrease in cardiac miR-30c expression levels in DCM rats
(3.73-fold), patients with DCM (2.9-fold) and in HG-treated
cardiomyocytes (3.5-fold) (35).
Further investigation indicated that miR-30c possessed binding
sites for the 3′-UTR and open reading frame (ORF) regions of
Cdc42 and Pak1, and modulated Cdc42 and
Pak1 expression levels in cardiomyocytes. In addition,
miR-30c overexpression decreased HG-induced upregulation of
Cdc42 and Pak1 and resulted in decreased expression
levels of hypertrophic marker, atrial natriuretic peptide and a
reduction in HG-treated cardiomyocyte cell size (35). These findings indicate that miR-30c
exerts an anti-hypertrophic effect by inhibiting Cdc42 and
Pak1 gene expression levels in DCM.
As a type of histone acetyl transferase,
transcriptional co-activator, p300 has been confirmed to
participate in the cardiomyocyte hypertrophy that is induced by
pro-hypertrophic stimuli, particularly hyperglycemia (29). Further investigations found that the
expression level of miR-150 was significantly reduced, whereas the
expression level of p300 was markedly elevated, concomitant
with cardiomyocyte hypertrophy, in the hypertrophic hearts of
diabetic rats and in neonatal rat cardiomyocytes exposed to high
levels of glucose (29). In addition,
a luciferase reporter activity assay indicated that miR-150
functioned directly with the 3′-UTR of p300 and miR-150
mimics prevented glucose-induced cardiomyocyte hypertrophy
(29). Thus, it was concluded that
miR-150 was important in p300-mediated cardiomyocyte hypertrophy in
response to hyperglycemia.
miR-373 has also been demonstrated to be involved in
the pathogenesis of hyperglycemia-induced cardiac hypertrophy. It
was found to be downregulated in heart samples of STZ-induced
diabetic mice, and exposure of neonatal rat cardiomyocytes to
glucose and transfection with miR-373 mimic demonstrated increased
expression levels of miR-373 and cell size, indicating a strong
involvement of miR-373 in glucose-induced cardiomyocyte hypertrophy
(36). In addition, the study revealed
that miR-373 was transcriptionally regulated by p38 MAPK and that
its anti-hypertrophic effects may be mediated by targeting the
hypertrophic protein, myocyte enhancer factor 2C (36).
Triglyceride accumulation and excess supply of
saturated fatty acids, such as palmitic acid, have been implicated
in the induction of cardiac hypertrophy in diabetes. miR-451
expression levels were observed to be significantly elevated in
diet-induced obesity (DIO) mouse hearts with hypertrophy and in
neonatal rat cardiomyocytes stimulated with palmitate (34). In addition, high-fat diet-induced
cardiac hypertrophy and contractile reserves were ameliorated in
cardiomyocyte-specific miR-451 knockout mice compared with the
control (34). As an important
component of the liver kinase B1 (LKB1)/adenosine monophosphate
activated protein kinase (AMPK) signaling pathway, calcium-binding
protein 39 (Cab39) was identified to be a direct miR-451 target in
neonatal rat cardiac myocytes. Further experiments indicated that
protein expression levels of Cab39 and phosphorylated AMPK were
increased and phosphorylated mammalian target of rapamycin (mTOR)
was reduced in cardiomyocyte-specific miR-451 knockout mouse hearts
compared with control mouse hearts, demonstrating that miR-451 was
involved in DCM via suppression of the LKB1/AMPK signaling pathway
(34).
miRs in myocardial fibrosis
Myocardial fibrosis is another main cause of DCM.
Abnormally elevated ECM deposition, in particular collagen
deposition, increases myocardial stiffness, leads to irreversible
tissue damage and finally results in myocardial fibrosis (16).
In addition to cardiac hypertrophy, miR-133a was
identified to be involved in the pathogenesis of diabetes-induced
myocardial fibrosis. The expression level of miR-133a was decreased
in the hearts of STZ-induced diabetic mice, accompanied by an
increase in the transcriptional co-activator, p300 and in major
markers of fibrosis (transforming growth factor-β1, connective
tissue growth factor, fibronectin and collagen 1 α1V), as well as
increased focal cardiac fibrosis, as measured by Masson's trichome
stain (28). Furthermore, miR-133a
overexpression prominently alleviates cardiac fibrosis as observed
by assessment of major fibrosis markers and microscopic
examination, indicating miR-133a as a potential therapeutic target
for combatting cardiac fibrosis (28).
The peripheral blood level of miR-21 has been
demonstrated as a biomarker for myocardial fibrosis (40). A recent study showed that miR-21 was
upregulated in rat cardiac fibroblasts in response to high levels
of glucose, accompanied by promoted fibroblast proliferation and
collagen synthesis (41). In addition,
dual specific phosphatase 8 (DUSP8) was identified to be a direct
target of miR-21; the expression of DUSP8 was suppressed by miR-21,
which promoted HG-induced cardiac fibrosis by affecting the
activity of the c-Jun N-terminal kinase/stress activated protein
kinase and p38 signaling pathways (41).
Thus, miR-133a and miR-21 are dysregulated by HG
stimulation, leading to myocardial fibrosis in DCM. Interventions
focusing on the expression levels of these miRs may result in novel
concepts for improving DCM remodeling.
miRs in cardiomyocyte apoptosis and
mitochondrial dysfunction
Various miRs have been demonstrated to be involved
in DCM-associated cardiomyocyte apoptosis and mitochondrial
dysfunction, including miR-34a, miR-1, miR-206, mi-195 and
mi-30d.
High levels of glucose may induce cardiomyocyte
apoptosis and thus contribute to the pathogenesis of DCM; miR-34a
was found to be involved in this process. Upregulation of miR-34a
expression levels and a decrease in the B cell leukemia/lymphoma 2
(Bcl-2) expression level were observed in the rat H9C2
cardiomyocyte cell line when exposed to HG, while apoptosis of H9C2
cells was significantly increased (42). Furthermore, treatment with miR-34a
mimics significantly decreased the Bcl-2 expression level
and promoted HG-induced apoptotic changes in H9C2 cells, whereas
treatment with an miR-34a inhibitor markedly increased the
Bcl-2 expression level and prevented H9C2 cell apoptosis,
indicating that miR-34a was critical in the HG-induced decrease of
the pro-apoptosis protein, Bcl-2 expression level and
subsequent cardiomyocyte apoptosis (42).
The molecular chaperone heat shock protein 60
(Hsp60) is an important anti-apoptotic protein, which may regulate
the Bcl-2 family. Reduced expression levels of the Hsp60 protein
were observed in the diabetic rat myocardium and HG-treated
neonatal rat ventricular cardiomyocytes, which was accompanied by
significant upregulation of miR-1 and miR-206 (43). Further studies then demonstrated that
rat miR-1 and miR-206 negatively regulated Hsp60 expression by
directly targeting the 3′-UTR of Hsp60 mRNA, and miR-1 and
miR-206 mediated their effects on H9C2 cell apoptosis via Hsp60
(43). These findings indicated that
miR-1 and miR-206 modulate the expression of their common target,
Hsp60 and consequently mediated HG-induced apoptosis of
cardiomyocytes.
In addition, miR-1 was found to mediate apoptosis of
HG-treated H9C2 cells through regulating another potential target,
IGF-1, which is proposed to be an anti-apoptosis factor (44). It was observed that H9C2 cells exposed
to HG levels exhibited increased miR-1 expression levels, decreased
mitochondrial membrane potential, increased cytochrome c release
and increased apoptosis; however, these consequences were detected
to be blocked by IGF-1 (44).
Another study demonstrated that the level of miR-195
expression was increased and expression levels of its target
proteins (Bcl-2 and sirtuin 1) were decreased in STZ-induced type 1
and db/db type 2 diabetic mouse hearts (45). Upregulation of miR-195 in diabetic
hearts was associated with oxidative stress, apoptosis, myocardial
hypertrophy and dysfunction, as well as a reduction in coronary
blood flow while silencing of miR-195 reduces oxidative damage,
apoptosis and hypertrophy, and restores coronary blood flow in
diabetic hearts, with a concurrent upregulation of Bcl-2 and
sirtuin 1, leading to an improvement in myocardial function
(45). This study validated the role
of miR-195 in promoting apoptosis in the DCM heart, as well as in
other pathophysiological changes (45).
Pyroptosis is pro-inflammatory programmed cell death
and it is another type of cell death that is different from
apoptosis or necrosis (46). HG may
induce cardiomyocyte pyroptosis and miR-30d was observed to be
involved in this process. It was revealed that miR-30d expression
was substantially increased in STZ-induced diabetic rats, as well
as in HG-treated cardiomyocytes (30).
Furthermore, upregulation of miR-30d promoted cardiomyocyte
pyroptosis in DCM by directly targeting forkhead box O3, which
resulted in suppression of the expression of its downstream
protein, apoptosis repressor with caspase recruitment domain and
upregulated expression of inflammatory molecules, including
caspase-1, interleukin (IL)-1β and IL-18, and finally led to
pyroptosis of cardiomyocyte (30).
Dysfunction of mitochondria also contributes to the
pathogenesis of DCM and miR-141 was found to participate in this
process. The expression level of miR-141 was significantly
upregulated in the hearts of STZ-induced diabetic mice.
Furthermore, through regulating its potential target, solute
carrier family 25 member 3, which provides inorganic phosphate to
the mitochondrial matrix and is essential for ATP production as an
inner mitochondrial membrane phosphate transporter, overexpression
of miR-141 was indicated to decrease inorganic phosphate transport
and exert functional implications for mitochondrial ATP production
(47).
miRs and other pathophysiological
processes of DCM
miRs are also involved in the pathogenesis of DCM
through participating in various pathophysiological processes, such
as myocardial electrical remodeling and epigenetic modification
(27,31).
A significant increase in the level of miR-301
expression and reduction of the voltage gated potassium channel,
Kv4.2 expression level were observed in the diabetic (db/db mice)
ventricles and miR-301 was validated to modulate Kv4.2 by direct
binding on its 3′-UTR (31). Kv4.2 is
important in maintaining the cardiac repolarization reserve, and
the depletion of repolarization reserve was further identified in
the diabetic hearts, elucidating that miR-301 mediated DCM by
regulating myocardial electrical remodeling (31).
DNA methylation is an important aspect of epigenetic
modification. In addition to its role in mediating myocardial
hypertrophy and fibrosis, miR-133a was found to contribute to
hyperglycemia-mediated DNA hypermethylation by regulating the
expression levels of DNA methyl transferases, which catalyze DNA
methylation. It was observed that the expression of miR-133a was
attenuated while DNA methyl transferase (Dnmt)-1 and −3b
were induced in Ins2+/− Akita hearts, and overexpression
of miR-133a inhibits, but silencing of miR-133a induces,
Dnmt-1, −3a and −3b, demonstrating the involvement of
miR-133a in the regulation of DNA methylation (27).
Conclusion
In conclusion, miRs are crucial in the pathogenesis
of DCM by regulating cardiomyocyte hypertrophy, myocardial
fibrosis, cardiomyocyte apoptosis, mitochondrial dysfunction,
myocardial electrical remodeling, epigenetic modification and
various other pathophysiological processes, as shown in Table I and Fig.
1. Furthermore, numerous studies have demonstrated that
interventions with the expression levels of associated miRNs may
improve the pathophysiological process of DCM, providing novel
insights into targets for the prevention and treatment of DCM
(28,29,34,35,45).
However, those studies were limited to the expression changes of
miRs in heart tissue samples. To the best of our knowledge,
circulating miRNs have not yet been identified to be specifically
dysregulated in DCM. Further studies and clinical observations are
required to identify circulating miRs as biomarkers for early
prediction and diagnosis of DCM.
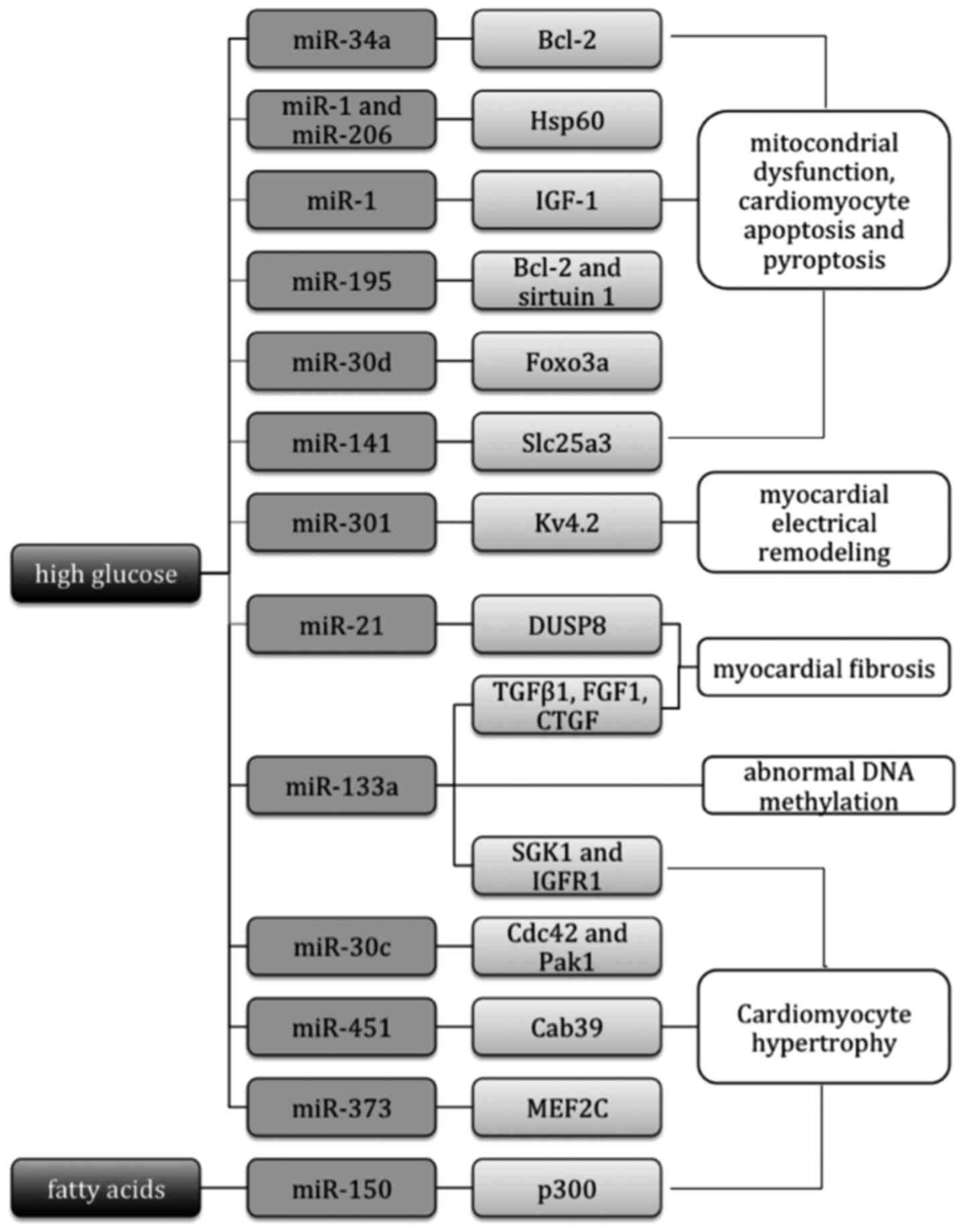 | Figure 1.miRs involved in the pathogenesis of
DCM. miRs participate in regulating cardiomyocyte hypertrophy,
myocardial fibrosis, cardiomyocyte apoptosis, mitochondrial
dysfunction, myocardial electrical remodeling and epigenetic
modification via their target genes. miRs, microRNAs; Bcl-2, B-cell
lymphoma 2; Hsp60, heat shock protein 60; IGF-1, insulin-like
growth factor-1; Foxo3a, forkhead box O3; Slc25a3, solute carrier
family 25 member 3; Kv4.2, voltage gate potassium channel 4.2;
DUSP8, dual specificity phosphatase 8; TGFβ1, transforming growth
factor-β1; FGF1, fibroblast growth factor 1; CTGF, connective
tissue growth factor; SGK1, serum and glucocorticoid-regulated
kinase 1; IGF1R, IGF-1 receptor; Cdc42, cell division control
protein 42 homolog; Pak1, p21-activated kinase 1; Cab39,
calcium-binding protein 39; MEF2C, myocyte enhancer factor 2C;
p300, transcriptional co-activator p300. |
 | Table I.miRs that are dysregulated in DCM and
their roles. |
Table I.
miRs that are dysregulated in DCM and
their roles.
| Author, year | miRs | Expression | Type of model
investigated | Potential
targets | Role in DCM | Refs. |
|---|
| Chavali et
al, 2012; | miR-133a | Downregulated | Cardiac tissues of
STZ-induced and Ins2+/− | SGK1 and
IGF1R; | Antihypertrophic
effect; regulating | (27,28,33) |
| Chen et al,
2014; and Feng et al, 2010 |
|
| Akita diabetic
mice; neonatal rat myocytes exposed to high levels of glucose | TGFβ1, FGF1,
CTGF | DNA methylation;
protection against myocardial fibrosis |
|
| Duan et al,
2013 | miR-150 | Downregulated | Hearts of STZ
diabetic SD rats; HG-treated primary neonatal rat
cardiomyocytes | p300 | Antihypertrophic
effect | (29) |
| Li et al,
2014 | miR-30d | Upregulated | Hearts of
STZ-induced diabetic rats and high glucose-treated
cardiomyocytes | Foxo3a | Promoting
pyroptosis | (30) |
| Panguluri et
al, 2013 | miR-301 | Upregulated | Right ventricle and
left ventricle of db/db mice | Kv4.2 | Inducing depletion
of repolarization reserve | (31) |
| Kuwabara et
al, 2015 | miR-451 | Upregulated | DIO mouse hearts;
neonatal rat cardiomyocytes stimulated with palmitate | Cab39 | Prohypertrophic
effect | (34) |
| Raut et al,
2015 | miR-30c | Downregulated | Hearts of Wistar
rats fed with HFD and low-dose STZ; cardiac tissue samples from
patients with DCM; HG-treated rat cardiomyocyte cell line
(H9C2) | Cdc42 and Pak1 | Antihypertrophic
effect | (35) |
| Shen et al,
2011 | miR-373 | Downregulated | Heart samples of
STZ-induced diabetic mice | MEF2C | Antihypertrophic
effect | (36) |
| Liu et al,
2014 | miR-21 | Upregulated | Rat cardiac
fibroblasts treated with a high level of glucose | DUSP8 | Promoting
myocardial fibrosis | (41) |
| Zhao et al,
2013 | miR-34a | Upregulated | High
glucose-treated rat cardiomyocyte H9C2 cell line | Bcl-2 | Promoting
apoptosis | (42) |
| Shan et al,
2010 | miR-1 and
miR-206 | Upregulated | Myocardium of
STZ-induced diabetic rat and high glucose-treated neonatal rat
ventricular cardiomyocytes | Hsp60 | Promoting
apoptosis | (43) |
| Yu et al,
2008 | miR-1 | Upregulated | H9C2 cells exposed
to high glucose | IGF-1 | Inducing
mitochondrial dysfunction, cytochrome c release and
apoptosis | (44) |
| Zheng et al,
2015 | miR-195 | Upregulated | Hearts of
STZ-induced type 1 and db/db type 2 diabetic mice; high
glucose-treated rat cardiomyocytes | Bcl-2 and sirtuin
1 | Promoting oxidative
stress, apoptosis, myocardial hypertrophy and dysfunction | (45) |
| Baseler et
al, 2012 | miR-141 | Upregulated | Hearts of
STZ-induced diabetic Friend Virus B mice | Slc25a3 | Decreasing
mitochondrial ATP production | (47) |
References
|
1
|
Guariguata L, Whiting DR, Hambleton I,
Beagley J, Linnenkamp U and Shaw JE: Global estimates of diabetes
prevalence for 2013 and projections for 2035. Diabetes Res Clin
Pract. 103:137–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wegner M, Neddermann D,
Piorunska-Stolzmann M and Jagodzinski PP: Role of epigenetic
mechanisms in the development of chronic complications of diabetes.
Diabetes Res Clin Pract. 105:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rawal S, Manning P and Katare R:
Cardiovascular microRNAs: As modulators and diagnostic biomarkers
of diabetic heart disease. Cardiovasc Diabetol. 13:442014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou Q, Lv D, Chen P, Xu T, Fu S, Li J and
Bei Y: MicroRNAs in diabetic cardiomyopathy and clinical
perspectives. Front Genet. 5:1852014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beckman JA, Creager MA and Libby P:
Diabetes and atherosclerosis: Epidemiology, pathophysiology, and
management. JAMA. 287:2570–2581. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chavali V, Tyagi SC and Mishra PK:
Predictors and prevention of diabetic cardiomyopathy. Diabetes
Metab Syndr Obes. 6:151–160. 2013.PubMed/NCBI
|
|
7
|
Hayat SA, Patel B, Khattar RS and Malik
RA: Diabetic cardiomyopathy: Mechanisms, diagnosis and treatment.
Clin Sci (Lond). 107:539–557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kannel WB and McGee DL: Diabetes and
glucose tolerance as risk factors for cardiovascular disease: The
Framingham study. Diabetes Care. 2:120–126. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Trachanas K, Sideris S, Aggeli C,
Poulidakis E, Gatzoulis K, Tousoulis D and Kallikazaros I: Diabetic
cardiomyopathy: From pathophysiology to treatment. Hellenic J
Cardiol. 55:411–421. 2014.PubMed/NCBI
|
|
11
|
Yilmaz S, Canpolat U, Aydogdu S and Abboud
HE: Diabetic Cardiomyopathy; Summary of 41 Years. Korean Circ J.
45:266–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Falcão-Pires I and Leite-Moreira AF:
Diabetic cardiomyopathy: Understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh GB, Sharma R and Khullar M:
Epigenetics and diabetic cardiomyopathy. Diabetes Res Clin Pract.
94:14–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Asrih M and Steffens S: Emerging role of
epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc
Pathol. 22:117–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu JW, Liu D, Cui KZ, Xu Y, Li YB, Sun YM
and Su Y: Recent advances in understanding the biochemical and
molecular mechanism of diabetic cardiomyopathy. Biochem Biophys Res
Commun. 427:441–443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huynh K, Bernardo BC, McMullen JR and
Ritchie RH: Diabetic cardiomyopathy: Mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bauersachs J and Thum T: Biogenesis and
regulation of cardiovascular microRNAs. Circ Res. 109:334–347.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Udali S, Guarini P, Moruzzi S, Choi SW and
Friso S: Cardiovascular epigenetics: From DNA methylation to
microRNAs. Mol Aspects Med. 34:883–901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fichtlscherer S, Zeiher AM, Dimmeler S and
Sessa WC: Circulating microRNAs: Biomarkers or mediators of
cardiovascular diseases? Arterioscler Thromb Vasc Biol.
31:2383–2390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fichtlscherer S, De Rosa S, Fox H,
Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Röxe T,
Müller-Ardogan M, et al: Circulating microRNAs in patients with
coronary artery disease. Circ Res. 107:677–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Figueira MF, Monnerat-Cahli G, Medei E,
Carvalho AB, Morales MM, Lamas ME, da Fonseca RN and Souza-Menezes
J: MicroRNAs: Potential therapeutic targets in diabetic
complications of the cardiovascular and renal systems. Acta Physiol
(Oxf). 211:491–500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Diao X, Shen E, Wang X and Hu B:
Differentially expressed microRNAs and their target genes in the
hearts of streptozotocin-induced diabetic mice. Mol Med Rep.
4:633–640. 2011.PubMed/NCBI
|
|
25
|
Rawal S, Ram TP, Coffey S, Williams MJ,
Saxena P, Bunton RW, Galvin IF and Katare R: Differential
expression pattern of cardiovascular microRNAs in the human type-2
diabetic heart with normal ejection fraction. Int J Cardiol.
202:40–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nandi SS, Duryee MJ, Shahshahan HR, Thiele
GM, Anderson DR and Mishra PK: Induction of autophagy markers is
associated with attenuation of miR-133a in diabetic heart failure
patients undergoing mechanical unloading. Am J Transl Res.
7:683–696. 2015.PubMed/NCBI
|
|
27
|
Chavali V, Tyagi SC and Mishra PK:
MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes.
Biochem Biophys Res Commun. 425:668–672. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen S, Puthanveetil P, Feng B, Matkovich
SJ, Dorn GW II and Chakrabarti S: Cardiac miR-133a overexpression
prevents early cardiac fibrosis in diabetes. J Cell Mol Med.
18:415–421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duan Y, Zhou B, Su H, Liu Y and Du C:
miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by
targeting the transcriptional co-activator p300. Exp Cell Res.
319:173–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Du N, Zhang Q, Li J, Chen X, Liu X,
Hu Y, Qin W, Shen N, Xu C, et al: MicroRNA-30d regulates
cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic
cardiomyopathy. Cell Death Dis. 5:e14792014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Panguluri SK, Tur J, Chapalamadugu KC,
Katnik C, Cuevas J and Tipparaju SM: MicroRNA-301a mediated
regulation of Kv4.2 in diabetes: Identification of key modulators.
PLoS One. 8:e605452013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu M, Liu Y, Zhang B, Shi Y, Cui L and
Zhao X: Inhibiting microRNA-144 abates oxidative stress and reduces
apoptosis in hearts of streptozotocin-induced diabetic mice.
Cardiovasc Pathol. 24:375–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Feng B, Chen S, George B, Feng Q and
Chakrabarti S: miR133a regulates cardiomyocyte hypertrophy in
diabetes. Diabetes Metab Res Rev. 26:40–49. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuwabara Y, Horie T, Baba O, Watanabe S,
Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, et al:
MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and
high-fat diet-induced cardiac hypertrophy in mice through
suppression of the LKB1/AMPK pathway. Circ Res. 116:279–288. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Raut SK, Kumar A, Singh GB, Nahar U,
Sharma V, Mittal A, Sharma R and Khullar M: miR-30c Mediates
Upregulation of Cdc42 and Pak1 in Diabetic Cardiomyopathy.
Cardiovasc Ther. 33:89–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen E, Diao X, Wang X, Chen R and Hu B:
MicroRNAs involved in the mitogen-activated protein kinase cascades
pathway during glucose-induced cardiomyocyte hypertrophy. Am J
Pathol. 179:639–650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu N, Bezprozvannaya S, Williams AH, Qi
X, Richardson JA, Bassel-Duby R and Olson EN: MicroRNA-133a
regulates cardiomyocyte proliferation and suppresses smooth muscle
gene expression in the heart. Genes Dev. 22:3242–3254. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Barringhaus KG and Zamore PD: MicroRNAs:
Regulating a change of heart. Circulation. 119:2217–2224. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Carè A, Catalucci D, Felicetti F, Bonci D,
Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, et al:
MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13:613–618.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen
H, Dean DB and Zhang C: MicroRNA expression signature and
antisense-mediated depletion reveal an essential role of MicroRNA
in vascular neointimal lesion formation. Circ Res. 100:1579–1588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu S, Li W, Xu M, Huang H, Wang J and
Chen X: Micro-RNA 21Targets dual specific phosphatase 8 to promote
collagen synthesis in high glucose-treated primary cardiac
fibroblasts. Can J Cardiol. 30:1689–1699. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao F, Li B, Wei YZ, Zhou B, Wang H, Chen
M, Gan XD, Wang ZH and Xiong SX: MicroRNA-34a regulates high
glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ
Sci Technolog Med Sci. 33:834–839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shan ZX, Lin QX, Deng CY, Zhu JN, Mai LP,
Liu JL, Fu YH, Liu XY, Li YX, Zhang YY, et al: miR-1/miR-206
regulate Hsp60 expression contributing to glucose-mediated
apoptosis in cardiomyocytes. FEBS Lett. 584:3592–3600. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX,
Lin SG and Li Y: Glucose induces apoptosis of cardiomyocytes via
microRNA-1 and IGF-1. Biochem Biophys Res Commun. 376:548–552.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G,
Chen R, Li J, Fan GC, Lacefield JC, et al: Silencing of miR-195
reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia.
58:1949–1958. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Nomenclature Committee on Cell Death
2009: Classification of cell death: Recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baseler WA, Thapa D, Jagannathan R,
Dabkowski ER, Croston TL and Hollander JM: miR-141 as a regulator
of the mitochondrial phosphate carrier (Slc25a3) in the type 1
diabetic heart. Am J Physiol Cell Physiol. 303:C1244–C1251. 2012.
View Article : Google Scholar : PubMed/NCBI
|














