Introduction
Next-generation sequencing (NGS) has become a
powerful and widely used clinical tool for the screening of
mutations in hereditary disease and for the evaluation of driver
and passenger mutations in cancer (1,2). Whole exome
sequencing (WES), which targets protein coding regions of the
genome, is the preferred option for the finding of new causative
genetic variants in rare Mendelian disorders as well as the
identification of genetic variants associated with individual types
of cancer (3,4). The Ion Torrent NGS platform implements a
semiconductor-based sequencing technology, the underlying principle
of which is the non-optical detection of hydrogen ions released
from the sequential addition of deoxynucleotides to a
deoxyribonucleic acid (DNA) chain (5).
The Ion Proton System, with 15 Gb output per run, and the AmpliSeq
Exome kit, which performs target enrichment of the entire human
exome by multiplex-polymerase chain reaction (PCR) amplification,
enable researchers to examine exomes rapidly and with low cost.
However, sequencing errors remain one of the main
obstacles in the identification of causative genetic variants
and/or mutations. Several patterns of sequencing error are
recognized based on the rationale of the NGS system. Compared to
the Illumina platforms, the Ion Proton platform has a high ratio of
false positives in the identification of small insertion and
deletion mutations (indel) but shows high accuracy in the
identification of single nucleotide variant (SNV) (6–8). Considering
that the vast majority of mutations (>90%) in human genome
present as SNVs, and that clinical practice necessitates rapid
sequencing at an acceptable cost for relatively few samples, the
Ion Proton System is an attractive option for clinical exome
evaluation. Therefore, information regarding SNV sequencing error
(such as frequency and/or etiologic character) is important, as it
has the potential to improve diagnostic accuracy and may avoid
waste in the clinical decision process.
In the present study, we describe the incidence and
characteristics of sequencing errors during WES with the Ion Proton
System, confirmed by Sanger sequencing.
Materials and methods
Overview
Two consanguineous patients were selected for WES.
Identified SNVs were validated with Sanger sequencing.
Ethics statement
The present study was approved by the Institute of
Biomedical Research and Innovation Hospitals Institutional Review
Board. All the patients provided written informed consent. The
study was conducted in accordance with the ethical principles of
the Declaration of Helsinki.
Blood samples and DNA isolation
The blood samples used in the study were collected
from the Institute of Biomedical Research and Innovation Hospital
(Kobe, Japan). DNA was isolated from peripheral blood mononuclear
cells using the QIAamp DNA Blood Mini kit (Qiagen, Hilden,
Germany), as per the manufacturers instructions. We measured DNA
concentration with NanoDrop Lite Spectrophotometer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Ion Torrent Proton library preparation
and sequencing
An Ion Torrent adapter-ligated library was generated
following the manufacturers protocol (Ion AmpliSeq™ Exome RDY kit
PIv3, Rev. A.0; MAN0010084; Thermo Fisher Scientific, Inc.).
Briefly, 100 ng high-quality genomic DNA was used to prepare the
Ion AmpliSeq™ Exome capture library. Pooled amplicons were
end-repaired, and Ion Torrent adapters and amplicons were ligated
with DNA ligase. Following AMPure bead purification (Beckman
Coulter, Inc., Brea, CA, USA), the concentration and size of the
library were determined using the Applied Biosystems®
StepOne™ Real-Time PCR system and Ion Library TaqMan®
Quantitation kit (both from Thermo Fisher Scientific, Inc.).
Sample emulsion PCR, emulsion breaking, and
enrichment were performed using the Ion PI™ Hi-Q™ Chef 200 kit
(Thermo Fisher Scientific, Inc.), according to the manufacturers
instructions. An input concentration of one DNA template copy per
ion sphere particles (ISPs) was added to the emulsion PCR master
mix and the emulsion was generated using the Ion Chef™ System
(Thermo Fisher Scientific, Inc.). Template-positive ISPs were
enriched, sequencing was performed using Ion PI™ Chip kit v3 chips
on the Ion Torrent Proton, and barcoding was performed using the
Ion DNA Barcoding kit (Thermo Fisher Scientific, Inc.).
Variant calling
Data from the Proton runs were initially processed
using Ion Torrent platform-specific pipeline software, Torrent
Suite v4.0 (Thermo Fisher Scientific, Inc.) to generate sequence
reads, trim adapter sequences, filter, and remove poor
signal-profile reads. Initial variant calling from the Ion
AmpliSeq™ sequencing data was generated using Torrent Suite with a
plug-in ‘variant caller’ program. To eliminate erroneous base
calling, three filtering steps were used to generate final variant
calling. The first filter was set at an average depth of total
coverage of >50, an each variant coverage of >15, and
P<0.01. The second filter was employed by visually examining
mutations using Integrative Genomics Viewer software (http://www.broadinstitute.org/igv) or CLC
Genomics Workbench version 9.5.1 (Qiagen), as well as by filtering
out possible strand-specific errors (i.e., a mutation was detected
only in one, but not both, strands of DNA).
Results
A total of 26,260 SNVs were initially detected. We
randomly selected 98 SNVs to be validated by Sanger sequencing. Of
these, nine single nucleotide sequence errors were identified. A
previous study identified primer-related sequencing errors in the
Ion Torrent system (9). Of the 18
total primers (forward and reverse) of the exome capture kit we
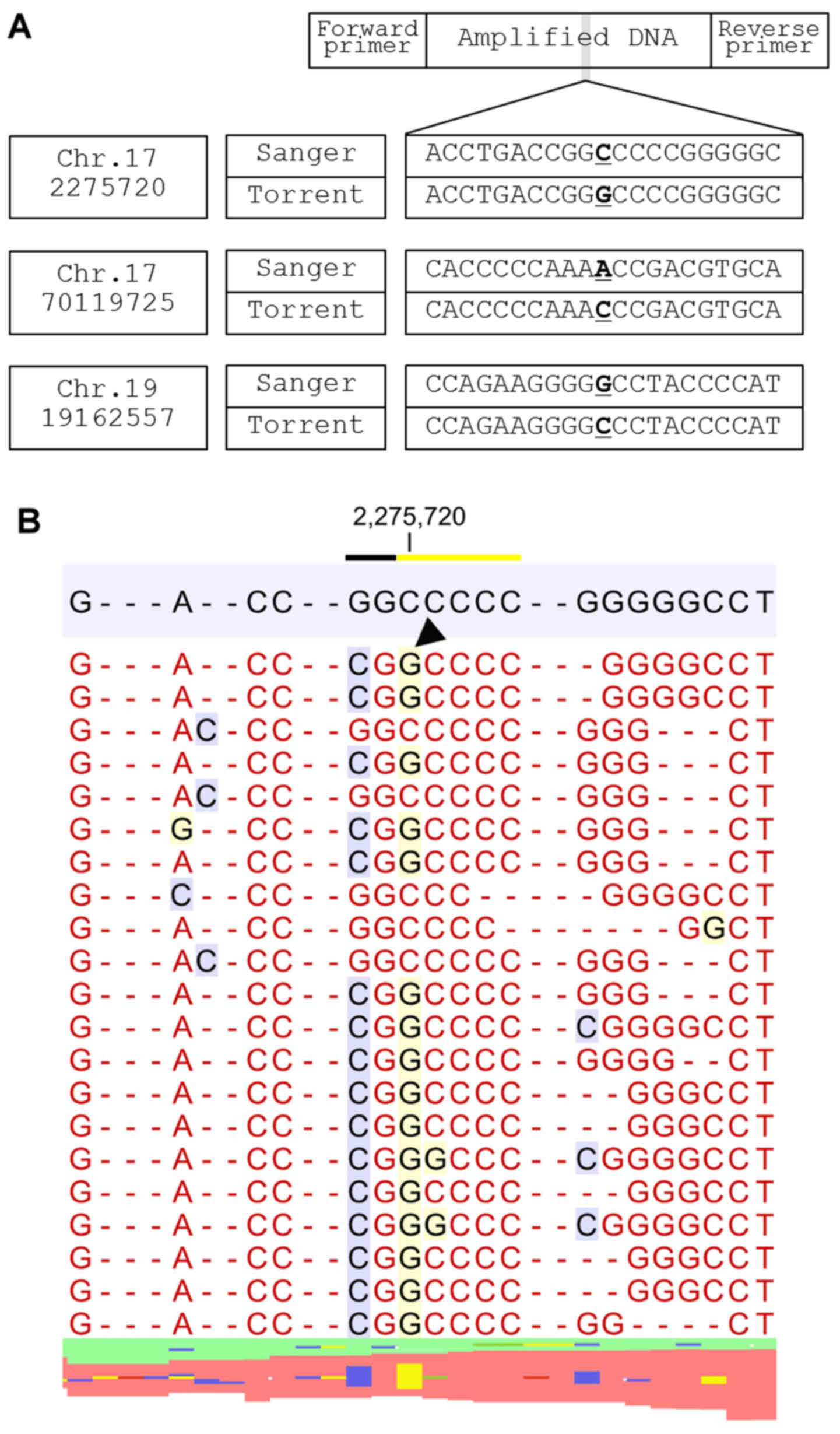
analyzed, one possible primer-related error was observed (Fig. 1).
The rest of the sequencing errors were unrelated to
primers and were classified into three groups. Group 1 sequencing
errors result from two homopolymer lesions, on both the 5′ and 3
sides, which cause a sequence error in the nucleotides of the
junctional area (Fig. 2). NGS showed
that loss of a nucleotide in one homopolymer area was accompanied
by gain of a nucleotide in the other homopolymer area. The
insertion locus was either at the immediate ends of the homopolymer
regions (e.g., where sequence AAAAGGGG is reported as AAAAAGGG) or
at a location a few nucleotides downstream of a homopolymer region
(e.g., where AAAAACCGT becomes AAAAACACGT and the left-side C is
masked, resulting in a sequencing error).
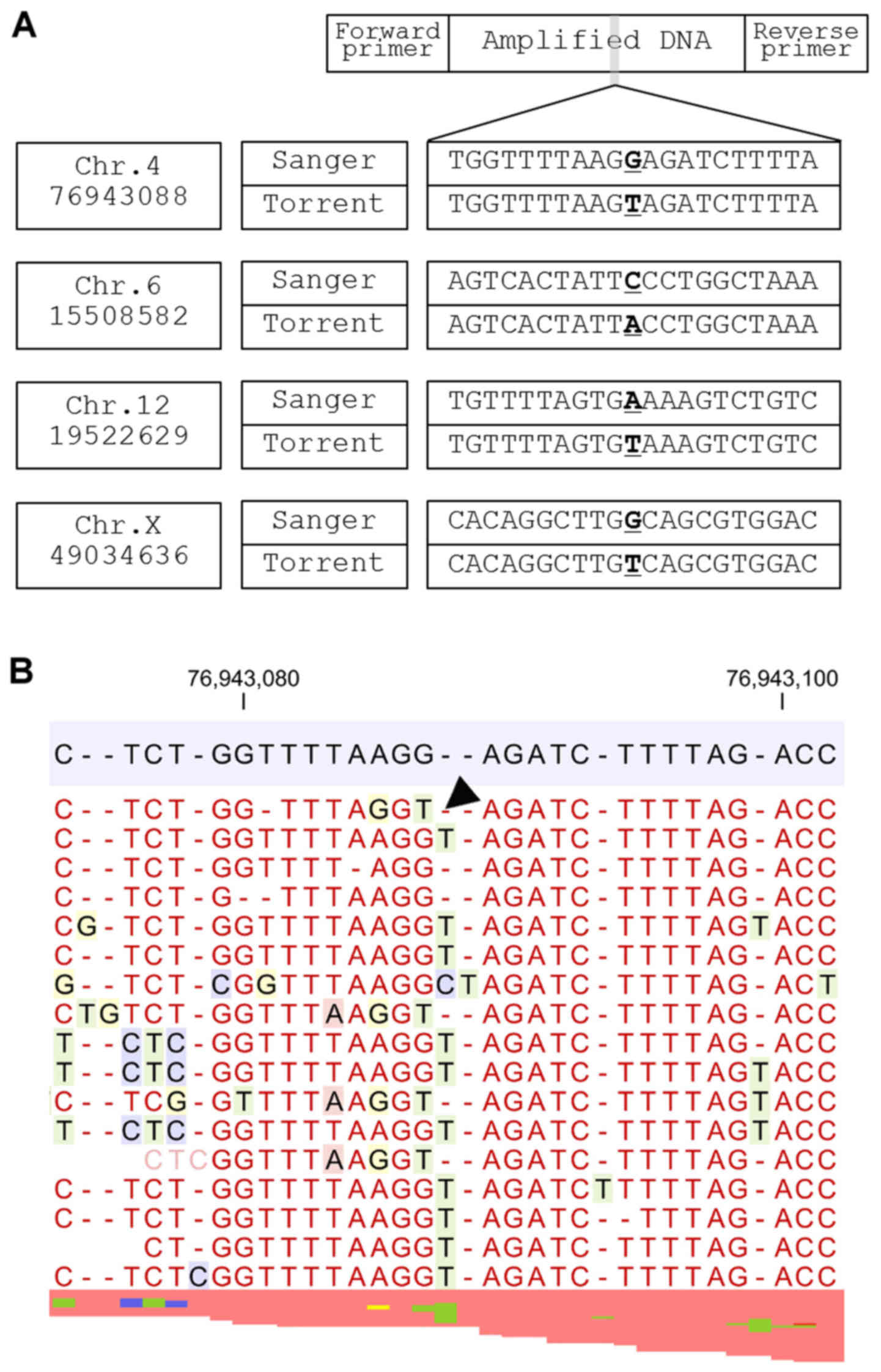
Group 2 sequencing errors occur when a nucleotide
lost from a homopolymer area is replaced by a nucleotide from
another homopolymer within 10 nucleotides of the observed sequence
error (Fig. 3). The neighboring
homopolymer lesion is considered to be the main cause of this
sequence error.
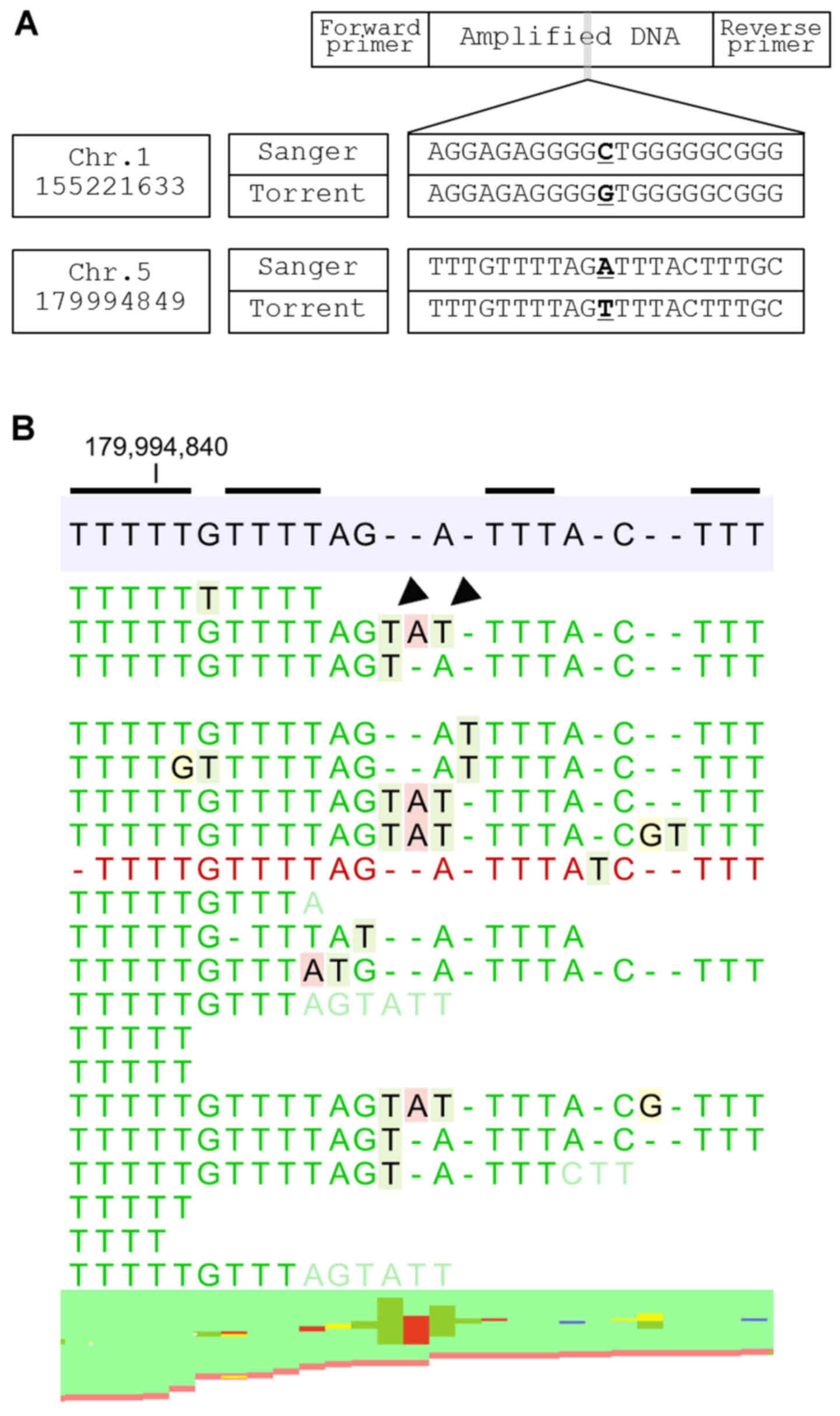
In Group 3, sequence error originates from the
‘elongation’ of a homopolymer lesion which may replace adjacent
nucleotides (e.g., GGGCTA becomes GGGGTA). Insertion of two
nucleotides, multiple homopolymers of which exist around the error,
are observed in the sequence-error site. These two nucleotides mask
the true nucleotide and a sequencing error occurs as a result
(Fig. 4). Notably, all of the
false-positive SNVs identified in the study are related to
homopolymer regions.
Discussion
The Ion Torrent system uses semiconductor sequencing
technology to detect the protons that are released as nucleotides
incorporated during DNA synthesis (5).
DNA fragments with specific adapter sequences are linked and then
clonally amplified by emulsion PCR on the surface of 3-µm beads.
These templated beads are loaded into proton-sensing microwells
fabricated on a silicon wafer, and sequencing is primed from a
specified location in the adapter sequence. A microwell is then
flooded with a single species of deoxyribonucleotide triphosphate
(dNTP). If the introduced dNTP is complementary to the leading
template nucleotide, it is incorporated into the growing
complementary strand (10). This
causes the release of a proton, which triggers an ion-sensitive
transistor sensor. If homopolymer repeats are present in the
template sequence, multiple dNTP molecules may be incorporated in a
single cycle, which leads to a corresponding number of released
hydrogens and a proportionally higher electronic signal. This
signal intensity is theoretically proportional to the number of
released nucleotides; however, miscount of the intensity can occur,
causing what is termed a homopolymer error. The majority of
homopolymer errors occur at the immediate ends of homopolymer
regions, where a sequence such as AAAAAAAA (an 8-nucleotide repeat)
is reported as AAAAAAAAA (a 9-nucleotide repeat) or AAAAAAA (a
7-nucleotide repeat). These errors can also occur at a location
several nucleotides downstream from a homopolymer region. In these
cases, a sequence such as AAAAAATGC is read as AAAAAATAGC.
Alternatively, a sequence of AAAAGTCG may be recognized as AAAAATCG
or AAACGTCG.
Sequencing errors remain a major challenge in the
analysis of SNVs using NGS platforms. Our results indicated that
SNVs located on junctional areas of two homopolymers (Group 1)
require confirmation with Sanger sequencing. SNVs originating from
homopolymer shortening or elongation (Groups 2 and 3, respectively)
are possibly false positives, and confirmation is necessary.
Several strategies are being investigated to avoid these
homopolymer sequencing errors. The Hi-Q enzyme was introduced in
2014 to enhance SNV and indel performance on the Ion PGM system and
later on the Ion Proton System. Internal tests indicated that the
Ion PGM system used with the Hi-Q kit achieved a 43% reduction of
indel errors in amplicon sequencing over the previous Ion PGM
sequencing enzyme. Another strategy is the development of software
for variant detection. Ion Reporter software is designed for
analyzing sequence data obtained from the Ion Torrent system. This
software is based on the algorithm of FreeBayes, a Bayesian genetic
variant detector designed to find small polymorphisms (11).
The present study revealed that the majority of the
SNV sequencing errors originate from homopolymer insertion/deletion
errors, are far more common in the Ion Torrent system than the
competitor Illumina system. Even when the abovementioned measures
are employed, the results obtained using the Ion Reporter System
should be interpreted with caution.
Acknowledgements
The present study was supported by JSPS KAKENHI
grant no. JP15K10017.
Glossary
Abbreviations
Abbreviations:
|
DNA
|
deoxyribonucleic acid
|
|
dNTP
|
deoxyribonucleotide triphosphate
|
|
ISPs
|
ion sphere particles
|
|
NGS
|
next-generation sequencing
|
|
PCR
|
polymerase chain reaction
|
|
SNV
|
single nucleotide variant
|
|
WES
|
whole exome sequencing
|
References
|
1
|
Pleasance ED, Cheetham RK, Stephens PJ,
McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR,
Bignell GR, et al: A comprehensive catalogue of somatic mutations
from a human cancer genome. Nature. 463:191–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Horak P, Frohling S and Glimm H:
Integrating next-generation sequencing into clinical oncology:
strategies, promises and pitfalls. ESMO Open. 1:e0000942016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bonnefond A, Durand E, Sand O, De Graeve
F, Gallina S, Busiah K, Lobbens S, Simon A, Bellanné-Chantelot C,
Létourneau L, et al: Molecular diagnosis of neonatal diabetes
mellitus using next-generation sequencing of the whole exome. PLoS
One. 5:e136302010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rothberg JM, Hinz W, Rearick TM, Schultz
J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M,
et al: An integrated semiconductor device enabling non-optical
genome sequencing. Nature. 475:348–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boland JF, Chung CC, Roberson D, Mitchell
J, Zhang X, Im KM, He J, Chanock SJ, Yeager M and Dean M: The new
sequencer on the block: Comparison of Life Technology's Proton
sequencer to an Illumina HiSeq for whole-exome sequencing. Human
genetics. 132:1153–1163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang G, Wang J, Yang J, Li W, Deng Y, Li
J, Huang J, Hu S and Zhang B: Comparison and evaluation of two
exome capture kits and sequencing platforms for variant calling.
BMC genomics. 16:5812015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Damiati E, Borsani G and Giacopuzzi E:
Amplicon-based semiconductor sequencing of human exomes:
performance evaluation and optimization strategies. Human genetics.
135:499–511. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McCall CM, Mosier S, Thiess M, Debeljak M,
Pallavajjala A, Beierl K, Deak KL, Datto MB, Gocke CD, Lin MT and
Eshleman JR: False positives in multiplex PCR-based next-generation
sequencing have unique signatures. JMD. 16:541–549. 2014.PubMed/NCBI
|
|
10
|
Thermo Fisher Scientific, Ion Torrent.
https://www.thermofisher.com/jp/en/home/brands/ion-torrent.htmlApril
10–2017
|
|
11
|
Garrison E and Marth G: Haplotype-based
variant detection from short-read sequencing.
arXiv.org>q-bio>arXiv:1207.3907. 2012
|