Introduction
Autosomal dominant optic atrophy (DOA) is a disorder
that results from the degeneration of the optic nerve fibers
(1). It is one of the most prevalent
forms of inherited optic neuropathies, which are genetic conditions
affecting the retinal ganglion cells whose axons constitute the
optic nerve (2). DOA is an important
cause of inherited visual failure and occurs equally among males
and females (1). Its prevalence is
estimated to be 1 per 30,000 worldwide with higher frequencies in
Denmark (1 per 10,000) due to a founder effect (3). It is generally diagnosed during childhood
and is characterized by loss of visual acuity, dyschromatopsia,
visual field defects and optic nerve pallor with optic disc
excavation (4).
The majority of DOA cases (80%) are isolated
(non-syndromic) while the remaining (20%) are syndromic (2). DOA that presents clinically as an
isolated optic neuropathy is caused by mutations in OPA1,
mitochondrial dynamin like GTPase (OPA1) (4), OPA3, outer mitochondrial membrane lipid
metabolism regulator (OPA3; also associated with cataracts)
(5), aconitase 2 (ACO2) (6) and transmembrane protein 126A (TMEM126A)
(7) genes, while syndromic DOA forms
show greater genetic heterogeneity (8). To date, 224 genes associated with optic
atrophy are listed in the Human Phenotype Ontology (HPO) database,
the majority presenting with neurological symptoms, such as ataxia,
mental retardation and spastic paraplegia (9). Increasing evidence also indicates that
other genes, in addition to yet unidentified genes, are involved
(10). Yet, despite these
advancements, more than half of DOA patients still await a genetic
diagnosis (11). This shortcoming may
be overcome using whole exome sequencing, which has the potential
to define the molecular diagnosis of patients presenting with a
negative result for mutations in the OPA1, OPA3, ACO2 and TMEM126A
genes.
In the current study, the exome of an Italian
patient affected by isolated DOA was analyzed. By combining exome
sequencing with phenotype-driven variant analysis, a heterozygous
mutation was identified in the AFG3 like matrix AAA peptidase
subunit 2 (AFG3L2) gene. Notably, the p.R468C (c.1402C>T)
mutation was recently described in a family exhibiting syndromic
DOA co-segregating with mild intellectual disability (11).
Patients and methods
Patient and clinical evaluation
A 19-year-old Italian male was admitted to the
Pediatric Low Vision Unit of the University Hospital of Padua
(Padua, Italy) with suspected optic atrophy. The patient had
experienced visual difficulties from the age of 6, including color
vision impairment and photophobia. When first examined, at 6 years
of age, ophthalmological investigations revealed a best corrected
visual acuity (BCVA) of 0.4 in each eye. Farnsworth testing
revealed dyschromatopsia and fundus examination exhibited bilateral
temporal optic nerve pallor. Intraocular pressure was within the
normal range (15 and 13 mmHg in the right and left eye,
respectively; normal range <18 mmHg). The anterior chamber of
the eyes exhibited no pathological features.
At the age of 13, photopic and scotopic
electroretinography tests did not reveal abnormal patterns, whereas
visual evoked potentials (VEPs) to pattern reversal stimuli
documented a significant decrease in the amplitude of the P100 wave
at 15′ and 30′ angular frequencies. As hereditary optic neuropathy
was suspected, genetic testing for OPA1 mutations, as well as for
the mitochondrial mutations associated with Leber neuropathy, were
performed with negative results.
BCVA worsened from 0.4 in each eye at age 6 to 0.2
in each eye at age 15. VEPs were repeated at the age of 17 and
exhibited considerable worsening of amplitude and waveform.
The individual, who is currently 21-years-old, did
not demonstrate further clinical deterioration. Furthermore,
multifocal electroretinogram and electrooculography did not detect
any alterations.
Whole exome sequencing
Whole blood (3 ml) was collected for exome analysis
subsequent to obtaining informed consent. DNA was extracted using
the Qiagen BioRobot DNA extraction kit (cat. no. 965162; Qiagen
Benelux B.V., Venlo, the Netherlands) according to the
manufacturer's instructions and quantified using Nanodrop spectral
analysis (Thermo Fischer Scientific, Inc., Waltham, MA, USA). DNA
fragmentation and degradation were evaluated by standard agarose
gel electrophoresis (100 V, 30 min, 1.5% agarose gel in
Tris-borate-EDTA buffer). DNA Library preparation and whole exon
enrichment were performed employing Agilent SureSelectXT and
Agilent SureSelect Clinical Research Exome (Agilent Technologies,
Inc., Santa Clara CA, USA). Library sequences were obtained using
the HiSeq2500 Illumina Sequencer (125-bp paired end sequence mode).
Bioinformatics analysis included the following: Next-generation
sequencing (NGS) reads mapping to whole genomes using the
Burrows-Wheeler Alignment tool (12)
with default parameters, polymerase chain reaction (PCR) duplicate
removal using Picard (http://picard.sourceforge.net), single nucleotide
polymorphisms and indel calling using the Genome Analysis Toolkit
(GATK) UnifiedGenotyper (13), variant
annotation using snpEff (http://snpeff.sourceforge.net) and false positive
variant filtration using the GATK VariantFiltration module.
Exome sequencing data and reads alignment analysis
had a mean depth of coverage (DoC) of 122.71×, with 95.7% of the
captured sequences displaying a mean DoC of 20×. The mean DoC
calculated for the mutated nucleotides was 69.0×.
An in silico multigene panel of 224 genes
listed in the HPO database, associated with optic atrophy, was used
to filter and select genetic variants obtained following exome
sequencing (gene list is available upon request). Variant
classification was performed in accordance with the recently
published guidelines from the American College of Medical Genetics
and Genomics (14).
Sanger sequencing of the variant
p.R468C (c.1402C>T) in the AGF3L2 gene
DNA (100 ng) was amplified using a standard PCR
procedure with a PCR mixture containing 2.5 µl 10X concentrated PCR
buffer, 0.7 µl 50 mM MgCl2 and 0.75 µl 10 mM deoxyribonucleotide
triphosphates (all from Solis BioDyne OÜ, Tartu, Estonia), 0.3 µl
of 100 µM forward primer (5′-TTTAGCGGTCGGAGATGAAC-3′) and 0.3 µl of
100 µM reverse primer (5′-CTGATGGTGCACTGTTGCTT-3′; Integrated DNA
Technologies, Coralville, IA, USA), and 0.5 µl 5 U/µl Hot Start DNA
Polymerase (Solis BioDyne OÜ). Thermocycling consisted of one cycle
of enzyme activation (at 95°C for 15 min), followed by 35 cycles of
DNA amplification (at 95°C for 45 sec, at 59°C for 45 sec and at
72°C for 1 min). PCR products were then separated by agarose gel
electrophoresis (100 V, 30 min, 1.5% agarose gel in
Tris-borate-EDTA buffer; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and purified with Invisorb Spin columns (Invitek Inc.,
Hayward, CA, USA). PCR-purified products were re-amplified with
terminating nucleotides using Big Dye Terminator v3.1 (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequencing analysis
was performed with an ABI Prism 3100 Avant automated sequencer
(Thermo Fisher Scientific, Inc.) equipped with 36-cm capillary
array filled with POP6 polymer (Thermo Fisher Scientific, Inc.).
Electropherograms were analyzed using Sequencing Analysis software
version 5.1.1 (Applied Biosystems; Thermo Fisher Scientific,
Inc.).
Results
Whole exome sequencing
Exome sequencing restricted to the 224 HPO genes
that were associated with isolated and syndromic optic atrophy
exhibited a mean DoC of 149.46×, with 99.00% of the captured
sequences displaying a DoC >10×, 98.07% >20× and 91.56%
>50×.
A total of 144 non-synonymous variants were detected
in the 224 HPO genes that were screened; of these, 19 had an allele
frequency <5% (data available upon request). Upon data analysis
and interpretation none of these variants were found to be of
pathogenic or potentially pathogenic significance. However, when
extending the genetic analysis across the whole exome, a
heterozygous p.R468C (c.1402C>T) mutation was identified in the
AFG3L2 gene. The p.R468C mutation is not reported in the public
accessible databases of human genetics (the Exome Aggregation
Consortium, Exome Variant Server or 1000 Genome Project). It is
located in an evolutionary conserved residue and computational
analysis predicts a potentially damaging effect on the resulting
protein (PholyPhen2 = 1.00) (phyloP-Vertebrate = 6.25/6.42)
(phyloP-Primate = 0.56/0.65).
Sanger sequencing. Sanger sequencing analysis
confirmed the presence of the p.R468C mutation in the patient and
excluded its presence in the patient's mother. The father was
untraceable; however, no signs of optic atrophy or cerebellar
dysfunctions were reported on his behalf.
Discussion
DOA is a highly genetically heterogeneous type of
optic neuropathy. Current diagnostic genetic testing approaches in
DOA searches for mutations in OPA1, OPA3, ACO2 and TMEM126A, with a
diagnostic success rate estimated to be ~70% (2). Gene sequencing is either performed by
conventional methods, such as Sanger sequencing or by targeted
capture NGS protocols. However, while single gene sequencing
(Sanger sequencing) is impractical for clinical practice, due to
time and budget constraints, the multigene targeted capture NGS
approach is limiting due to gene selection bias. In addition,
increasing genotype-phenotype associations indicates that other
genes, in addition to yet unidentified genes, are likely to be
implicated in DOA. Currently, the genetic testing approaches that
theoretically guarantee the greatest diagnostic success are whole
exome and whole genome sequencing.
In the current study, exome sequence analysis and
interpretation in the patient revealed that there were no
pathogenic or likely pathogenic mutations in the four genes
typically associated with isolated DOA. Subsequently, the analysis
was extended to include those genes listed in the HPO database
associated with optic atrophy (primarily syndromic) and again the
results were negative. Therefore, the whole exome was subsequently
analyzed using the phenotype-driven strategy for exome
prioritization of human Mendelian disease genes (15,16).
Notably, a heterozygous p.R468C (c.1402C>T) mutation was
identified in exon 11 of the AFG3L2 gene. This gene interacts with
the protein encoded by the SPG7 gene, which is one of the 224 genes
listed in the HPO database.
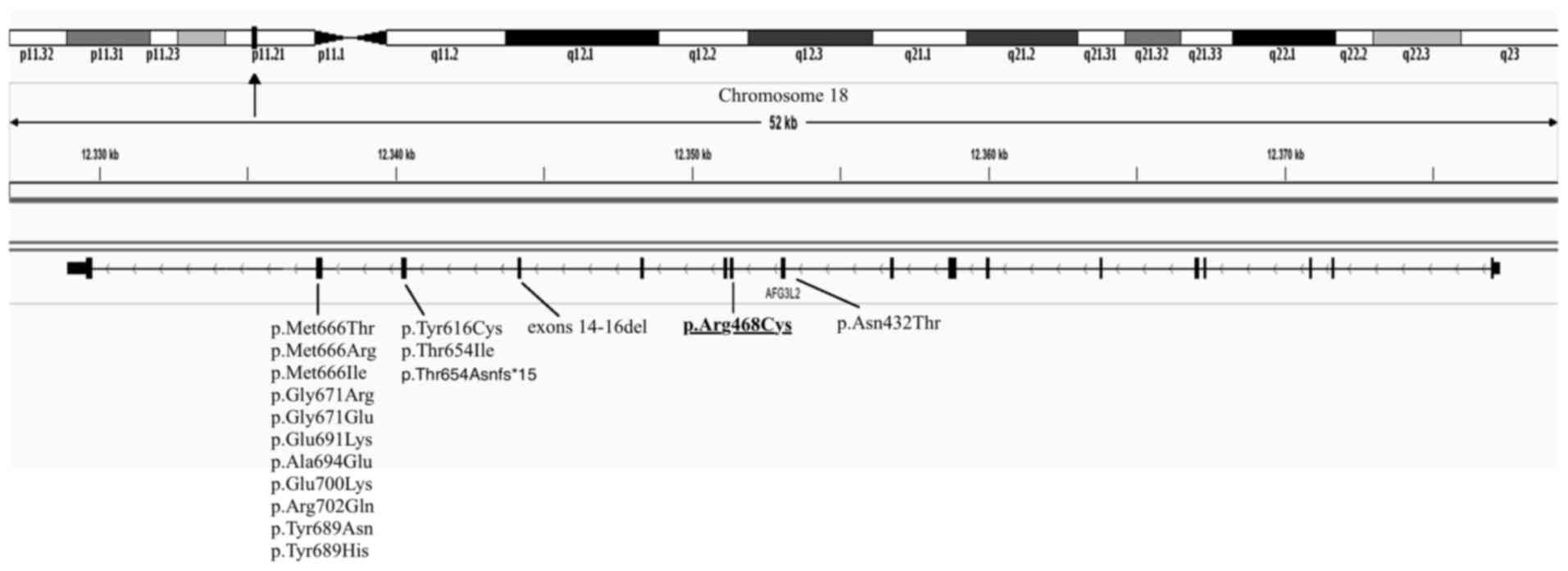
The AFG3L2 gene contains 17 exons spanning 48 kb and
maps to chromosome 18p11.21 (17). It
encodes a 797-amino acid protein localized in the mitochondria and
it is ubiquitously expressed in central and peripheral nerves
(17). The AFG3L2 protein assembles
with the homologous protein paraplegin to form large proteolytic
complexes responsible for mitochondrial protein quality control
(18,19). Functional studies in neurons
demonstrated a role of AFG3L2 in axonal development and activity of
mitochondrial respiratory complexes I and III (19). DOA is a mitochondrial disorder, thus
reinforcing the possibility of an association between AFG3L2 and
this particular neuropathy.
The p.R468C mutation in the AFG3L2 gene observed in
the patient has previously been described in two patients (a father
and son) with DOA and mild intellectual disability (11). The authors, however, did not exclude
the possibility of a fortuitous association between the two
phenotypes (11). In the present
study, the proband exhibited no signs of intellectual disability.
However, as the p.R468C mutation in the AFG3L2 gene is particularly
rare, and given the limited available information in previous
studies, interfamilial phenotype differences and/or penetrance
cannot be excluded.
Currently, the majority of mutations in the AFG3L2
gene have been reported to be associated with autosomal dominant
spinocerebellar ataxia 28 (17,20) and
autosomal recessive spastic ataxia 5 (21,22) (MIM no.
604581). The patient in the present study currently exhibits
isolated DOA with no signs of ataxia. This indicates that different
mutations in various locations in the AFG3L2 gene may lead to
different clinical phenotypes. Furthermore, the p.R468C mutation is
located in exon 11, while all known ataxia-causing mutations,
except one, are located in exons 15 and 16 of AFG3L2 (23,24)
(Fig. 1). However, the mutation may be
mild and/or cause late-onset ataxia or extra-ocular signs (11), thus requiring ongoing surveillance of
the patient.
In conclusion, the current study identified, to the
best of our knowledge, the first case of non-syndromic isolated
optic atrophy potentially associated with a heterozygous pathogenic
mutation in the AFG3L2 gene. Screening for mutations in this gene
may facilitate with characterizing the genotype-phenotype
association of AFG3L2 genetic variations in those patients without
a genetic diagnosis of DOA. Using whole exome sequencing in
clinical practice will further identify the disease aetiology of
DOA, ideally facilitating genetic diagnosis, as well as the design
of future treatment strategies.
References
|
1
|
Yu-Wai-Man P, Griffiths PG, Burke A,
Sellar PW, Clarke MP, Gnanaraj L, Ah-Kine D, Hudson G, Czermin B,
Taylor RW, et al: The prevalence and natural history of dominant
optic atrophy due to OPA1 mutations. Ophthalmology.
117:1538–1546.e1. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lenaers G, Hamel C, Delettre C,
Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P and Milea D:
Dominant optic atrophy. Orphanet J Rare Dis. 7:462012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiselton DL, Alexander C, Morris A,
Brooks S, Rosenberg T, Eiberg H, Kjer B, Kjer P, Bhattacharya SS
and Votruba M: A frameshift mutation in exon 28 of the OPA1 gene
explains the high prevalence of dominant optic atrophy in the
Danish population: Evidence for a founder effect. Hum Genet.
109:498–502. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferré M, Caignard A, Milea D, Leruez S,
Cassereau J, Chevrollier A, Amati-Bonneau P, Verny C, Bonneau D,
Procaccio V and Reynier P: Improved locus-specific database for
OPA1 mutations allows inclusion of advanced clinical data. Hum
Mutat. 36:20–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reynier P, Amati-Bonneau P, Verny C,
Olichon A, Simard G, Guichet A, Bonnemains C, Malecaze F, Malinge
MC, Pelletier JB, et al: OPA3 gene mutations responsible for
autosomal dominant optic atrophy and cataract. J Med Genet.
41:e110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Metodiev MD, Gerber S, Hubert L, Delahodde
A, Chretien D, Gérard X, Amati-Bonneau P, Giacomotto MC, Boddaert
N, Kaminska A, et al: Mutations in the tricarboxylic acid cycle
enzyme, aconitase 2, cause either isolated or syndromic optic
neuropathy with encephalopathy and cerebellar atrophy. J Med Genet.
51:834–838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hanein S, Perrault I, Roche O, Gerber S,
Khadom N, Rio M, Boddaert N, Jean-Pierre M, Brahimi N, Serre V, et
al: TMEM126A, encoding a mitochondrial protein, is mutated in
autosomal-recessive nonsyndromic optic atrophy. Am J Hum Genet.
84:493–498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maresca A, la Morgia C, Caporali L,
Valentino ML and Carelli V: The optic nerve: A ‘mito-window’ on
mitochondrial neurodegeneration. Mol Cell Neurosci. 55:62–76. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Optic atrophy. http://compbio.charite.de/hpoweb/showterm?id=HP:0000648July
18–2016
|
|
10
|
Grenier J, Meunier I, Daien V, Baudoin C,
Halloy F, Bocquet B, Blanchet C, Delettre C, Esmenjaud E, Roubertie
A, et al: WFS1 in optic neuropathies: Mutation findings in
nonsyndromic optic atrophy and assessment of clinical severity.
Ophthalmology. 123:1989–1998. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Charif M, Roubertie A, Salime S, Mamouni
S, Goizet C, Hamel CP and Lenaers G: A novel mutation of AFG3L2
might cause dominant optic atrophy in patients with mild
intellectual disability. Front Genet. 6:3112015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 11:11.10.1–11.10.33. 2013.
|
|
14
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zemojtel T, Köhler S, Mackenroth L, Jäger
M, Hecht J, Krawitz P, Graul-Neumann L, Doelken S, Ehmke N,
Spielmann M, et al: Effective diagnosis of genetic disease by
computational phenotype analysis of the disease-associated genome.
Sci Transl Med. 6:252ra1232014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smedley D and Robinson PN:
Phenotype-driven strategies for exome prioritization of human
Mendelian disease genes. Genome Med. 7:812015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Di Bella D, Lazzaro F, Brusco A, Plumari
M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali
M, et al: Mutations in the mitochondrial protease gene AFG3L2 cause
dominant hereditary ataxia SCA28. Nat Genet. 42:313–321. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zühlke C, Mikat B, Timmann D, Wieczorek D,
Gillessen-Kaesbach G and Bürk K: Spinocerebellar ataxia 28: A novel
AFG3L2 mutation in a German family with young onset, slow
progression and saccadic slowing. Cerebellum Ataxias. 2:192015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maltecca F, Aghaie A, Schroeder DG,
Cassina L, Taylor BA, Phillips SJ, Malaguti M, Previtali S, Guénet
JL, Quattrini A, et al: The mitochondrial protease AFG3L2 is
essential for axonal development. J Neurosci. 28:2827–2836. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cagnoli C, Stevanin G, Brussino A,
Barberis M, Mancini C, Margolis RL, Holmes SE, Nobili M, Forlani S,
Padovan S, et al: Missense mutations in the AFG3L2 proteolytic
domain account for ~1.5% of European autosomal dominant cerebellar
ataxias. Hum Mutat. 31:1117–1124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pierson TM, Adams D, Bonn F, Martinelli P,
Cherukuri PF, Teer JK, Hansen NF and Cruz P: Mullikin For The Nisc
Comparative Sequencing Program JC Blakesley RW et al. Whole-exome
sequencing identifies homozygous AFG3L2 mutations in a spastic
ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases.
PLoS Genet. 7:e10023252011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muona M, Berkovic SF, Dibbens LM, Oliver
KL, Maljevic S, Bayly MA, Joensuu T, Canafoglia L, Franceschetti S,
Michelucci R, et al: A recurrent de novo mutation in KCNC1 causes
progressive myoclonus epilepsy. Nat Genet. 47:39–46. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Löbbe AM, Kang JS, Hilker R, Hackstein H,
Müller U and Nolte D: A novel missense mutation in AFG3L2
associated with late onset and slow progression of spinocerebellar
ataxia type 28. J Mol Neurosci. 52:493–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Musova Z, Kaiserova M, Kriegova E,
Fillerova R, Vasovcak P, Santava A, Mensikova K, Zumrova A,
Krepelova A, Sedlacek Z and Kanovsky P: A novel frameshift mutation
in the AFG3L2 gene in a patient with spinocerebellar ataxia.
Cerebellum. 13:331–337. 2014. View Article : Google Scholar : PubMed/NCBI
|