Introduction
Chromosomal imbalances are frequently associated
with dysmorphism, congenital malformations and psychomotor
impairment (1). Aneuploidies
involving human chromosome 21 (Hsa21) are the most prevalent
chromosomal abnormalities in humans, with down syndrome (DS)
describing the presence of an extra copy of Hsa21, occurring in ~1
of 733 live births (2), and monosomy
21 being very rare (<0.01%) resulting in mortality in
utero or shortly following birth (3). Partial monosomy and trisomy are
difficult to detect with conventional cytogenetic techniques
(1). Novel technologies, including
high-resolution microarray comparative genomic hybridization
(aCGH), quantitative polymerase chain reaction (qPCR) and
fluorescent in situ hybridization (FISH) allow identifying
chromosomal imbalances and aid correlating geno- and phenotypes in
patients (1). This information may be
used in genetic counseling, which is an aim of the present
study.
Partial deletion of Hsa21 is more frequent than
complete monosomy and patients have increased survival expectancy
and are heterogeneous regarding their phenotypic severity depending
on the position and size of the deleted region (4-7).
There are >45 cases reported in literature. Lyle et al
(6) differentiate three regions
associated with various levels of clinical severity, ranging from
moderate to not compatible with life. The deleted region described
in this study was consistent with that Roberson et al
(7) reported as the most frequently
altered and with the mildest phenotypic expression.
The occurrence of segmental trisomy 21, with or
without association to DS, was first reported by Ilbery et
al (8) in 1961. Approximately 200
cases have been reported in literature until 2016 compared with an
estimated 5.8 million patients with DS worldwide (9). It has been demonstrated that only part
of the Hsa21 is involved in the pathogenesis of DS (9). Pelleri et al (9) reported a highly restricted DS critical
region of 34 kb, with no known genes located in it and it was
identified as the minimal region duplicated patients with DS and
absent healthy subjects.
The present study described a patient carrying a
21q22.3 deletion and duplication as detected by aCGH array.
Symptoms included facial dysmorphism, hypotonia, short stature,
clinodactyly, learning impairment, autism spectrum disorder (ASD),
anxiety and depression. To the best of our knowledge this is the
first case reporting these two copy number variants (CNVs) to occur
simultaneously and these regions were the shortest described so
far. The analysis of these partial aneuploidies aimed to correlate
clinical features of the phenotype to this specific Hsa21
region.
Clinical report
Patient details
A full-term male born to unrelated, healthy parents
following an uncomplicated pregnancy was reported. The patient
first presented at the Developmental Assessment Unit of the 2nd
Department of Pediatrics, ‘P. & A. Kyriakou’ Children's
Hospital (Athens, Greece) in October 2015. The patient was 10 years
at the time of recruitment and he was the first child of the
family. The patient was delivered by cesarean section with 3,150 g
birth weight (25th percentile), 50 cm length (50th percentile) and
34 cm head circumference (HC; 25th percentile). No abnormalities or
complications were recorded in the perinatal history. Developmental
milestones were reported normal; the patient sat unsupported at 7
months, walked unaided at 14 months and started speaking at 13
months.
At 10 years old, the patient was referred for full
developmental assessment due to learning difficulties and
behavioral problems at school, affecting social relationships with
peers. Between 10-12 years, the patient received psychological
treatment. The patient exhibited a shy demeanor, short statue,
microcephaly and minor dysmorphic facial and body features,
including long philtrum, frontal bossing, almond shaped eyes,
auricle abnormalities, nipples widely spaced and clinodactyly of
the fifth finger. At the time of recruitment, the following details
were recorded: Height, 130 cm (3rd percentile); weight, 33 kg (25th
percentile); and HC, 51 cm (<3rd percentile). The intellectual
abilities were tested using the Wechsler intelligence scale for
children (WISC III) (10) and a
difference between verbal and performance skills was determined.
The verbal score was at upper limit (verbal IQ=111) and the
performance score was at lower end (performance IQ=83) of the mean
IQ range (IQ, 80-119) (10). Child
psychiatric evaluation exhibited severe anxiety and depression
levels.
At 12 years, a psychiatric evaluation diagnosed
autistic spectrum disorder (ASD) according to diagnostic and
statistical manual of mental disorders (DSM)-5 criteria (11). Combined educational and pharmaceutical
treatment followed (risperidone, 0.25 mg twice daily). Clinical
neurological examination revealed global hypotonia without focal
neurological signs and microcephaly. Laboratory investigations,
including audiological, visual, biochemical, metabolic, endocrine
(thyroid, growth hormone, luteinizing hormone, follicle stimulating
hormone, adrenocorticotropic hormone and prolactin), bone age,
kidney/liver ultra sound, triplex ultrasound, brain magnetic
resonance imaging and electroencephalogram were normal.
Materials and methods
Metaphase chromosomes were obtained from
phytohemagglutinin-stimulated peripheral blood lymphocytes and
high-resolution (thymidine treatment) G-banding karyotype analysis
was performed using standard procedures (12).
aCGH was performed by normalizing the sample against
a male human reference commercial DNA sample using an aCGH platform
that includes 60,000 oligonucleotides as described elsewhere
(9), distributed across the entire
genome (cat. no. 5190-3796; Agilent Technologies, Inc., Santa
Clara, CA, USA). Statistical analyses estimating the number of
copies were performed using Cytogenomics v4.0 (Agilent
Technologies, Inc.) with a window of 0.5 Mb and A=6. If copy number
changes affected ≥5 consecutive probes with identically oriented
change, these were considered as CNVs. For the majority of the
genome, the mean genomic power of resolution was 200 kb. The
genomic coordinates were listed according to genomic build
GRCh37/hg19 (genome.ucsc.edu).
Results
The cytogenetic analysis revealed a normal male
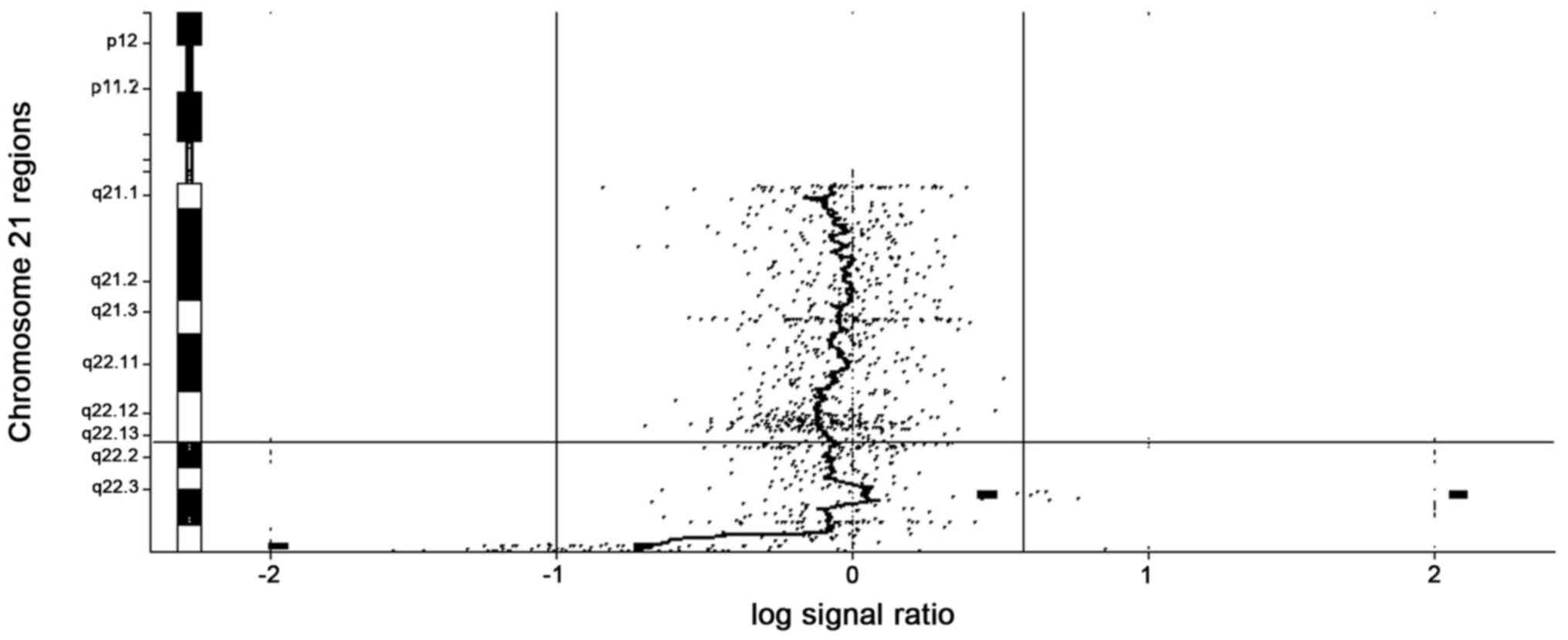
karyotype (46,XY). aCGH analysis detected the loss of ~650 kb and a
copy number gain of ~760 kb in the 21q22.3 region (Fig. 1). These alterations affected 13 and 11
genes, respectively. The deleted segment was mapped at the
chr21:47,439,416-48,090,317 region and the duplicated segment was
mapped at the chr21:42,710,862-43,473,699 region. The deletion
spanned ten OMIM genes and was one of the shortest described in the
literature so far (Table I). The
duplicated region described in the present study covers the
proximal ~760 kb of the 21q22.3 region and includes eight online
Mendelian inheritance in man (OMIM) genes (Table II).
 | Table IDeleted OMIM genes determined for the
10-year-old male patient. |
Table I
Deleted OMIM genes determined for the
10-year-old male patient.
| Gene | OMIM reference |
|---|
| COL26A2 | 120240 |
| FTCD | 606806 |
|
C21orf56 | 612412 |
| LSS | 600909 |
| MCM3AP | 603294 |
| YBEY | 617461 |
| PCNT | 605925 |
| DIP2A | 607711 |
| S100B | 176990 |
| HRMT1L | 601961 |
| Table IIDuplicated OMIM genes determined for
the 10-year-old male patient. |
Table II
Duplicated OMIM genes determined for
the 10-year-old male patient.
| Gene | OMIM reference |
|---|
| FAM3B | 608617 |
| MX2 | 147890 |
| MX1 | 147150 |
| TMPRSS2 | 602060 |
| RIPK4 | 605706 |
| PRDM15 | 617692 |
| C2CD2 | 617581 |
| ZBTB21 | 616485 |
Discussion
The study of chromosomal abnormalities is the most
commonly used tool for the definition of the morbidity map of the
human genome (1). In this respect, it
is of fundamental importance to study small deletions/duplications
(<1 Mb), which may be characterized with high accuracy due to
the introduction of aCGH (13).
The current study presented the case of a 12-year
old male patient with normal perinatal and infant development. By
10 years, the patient exhibited learning difficulties and
behavioral problems, resulting in a referral for full developmental
assessment that revealed short stature, microcephaly, hypotonia and
minor facial and trunk dysmorphic features. At 10 years, a
psychiatric evaluation suggested severe anxiety and depression that
strongly conditioned social relations. On an intellectual level,
there was a discrepancy between verbal and performance skills. At
12 years, a psychiatric evaluation diagnosed ASD according to DSM-V
criteria. Using aCGH, a duplication of ~760 kb and a deletion of
~650 kb in the 21q22.3 region were identified.
Although DS is the most common chromosomal
abnormality, 125 cases describing segmental trisomy of Hsa21 have
been published in the literature so far (9). Few studies identified the chromosomal
region of interest as 21q22 (6,9). Over
forty cases of partial trisomy of Hsa21 that include the 21q22.3
region have been described in the literature to date (4-7).
A comparison between the present study and the literature has
proven difficult due to variations in the size of the duplication
and the presence of other chromosomal aberrations. Additionally,
certain clinical characteristics described in the present study,
including short stature, microcephaly, hypotonia and clinodactyly
of fifth finger are associated with DS, but are also common to
other chromosomal abnormalities. Therefore, it was not possible to
attribute the characteristics specifically to the partial trisomy
21.
There is growing evidence that trisomy of the
21q22.3 segment exhibits no clinical significance (14). A terminal duplication of ~5 Mb of the
21q22.3 region was observed in a 2-year-old male exhibiting no
pathological phenotype (15). An
interstitial duplication of 4.4 Mb of the 21q22-22.3 region was
detected in a 2-year-old healthy female and her 37-year-old healthy
mother (16). In a large subtelomere
FISH analysis, two cases presented with a 21q deletion and
exhibited no clinical signs of disease (17). Therefore, the pathological
characteristics observed in the patient of the present study were
suggested to be attributed to a combination of the partial trisomy
and the terminal deletion of the distal region of 21q22.3.
Comparable combinations of an inverted duplication adjacent to a
terminal deletion have been described for chromosome arms 2q, 4p,
5p, 6q, 8p, 9p, 10q, 13q, 15q, 18p, 18q and 22q (18). The mechanisms proposed for the complex
rearrangement are based on non-allelic homologous recombination,
U-type exchange and telomere capture (18,19). For
the present case, the underlying mechanisms were not identified and
it remains to be investigated whether the duplication is inverted.
Future studies may include the use of FISH aiding to identify a
potential inversion, and sequencing of the regions in proximity to
the breakpoints may reveal the mechanisms of rearrangement.
Lyle et al (6)
proposed the classification of partial 21q monosomy into three
groups: i) Deletions starting from the centromere up to 32.3 Mb;
ii) deletions in the 32.2-37.1 Mb region; and iii) deletions from
37 Mb to the telomere. The first two groups of 21q monosomy are
associated with clinically severe phenotypes, while the third group
exhibits moderate phenotypes (6).
Clinical characteristics observed in the present patient frequently
reported in the literature for partial Hsa21q monosomy included
microcephaly and short stature with growth delay.
A chromosome 21 ring carrying a deletion that
includes the genes COL6A2 and S100B is propagated in
a family. Four individuals from three generations with monosomy
were described and patients were diagnosed with a medium to severe
growth retardation, short stature and microcephaly (20). McGinniss et al (21) described 13 patients with Hsa21 rings;
five were clinically healthy and had a deletion with two
breakpoints between COL6A1 and S100B. Mental
retardation of mild grade, growth retardation, short stature and
microcephaly were observed in a patient with a longer deletion
including the COL6A1 gene. Roberson et al (7) presented ten novel cases of partial 21q
monosomy, including a 4-year-old male with a de novo 5.68 Mb
21q22.3 terminal deletion presenting moderate mental retardation,
strabismus, normal growth and an absence of dysmorphism. The Hsa21
monosomal region observed in the present case included 10 OMIM
genes and to date no genes with pathological phenotype have been
mapped in this region.
The patient of the present study exhibited anxiety
and severe depression that affected academic performance and
relationships with peers at 10 years. The difference between verbal
and performance skills evaluated using the WISC-III test was
attributed to these symptoms. At 12 years, a psychiatric evaluation
diagnosed ASD according to DSM-V criteria. Other family members
were not diagnosed with mood disorders. Anxiety and depression are
clinical signs rarely reported in patients with chromosomal
abnormalities. This may be due to geneticists consulting on
pediatric cases and follow-up to when psychiatric disorders
manifest are rare. Psychiatrists often do not consider genetic
syndromes in the diagnosis of their patients (22). However, depression is difficult to
diagnose during childhood as it may be masked by major phenotypes,
including even mild mental retardation (23). Therefore, anxiety and depression may
be underestimated with respect to actual prevalence. A recent study
has suggested that occurrence of depression and anxiety is frequent
(20%) in individuals with ASD and is associated with higher IQs and
fewer ASD symptoms (24). Two
associated studies have highlighted a susceptibility locus for
bipolar depression, which maps to the 21q22 region (25,26). Major
depression was observed in 16- and 18-year-old brothers with a
terminal deletion of ~200 kb in the 21q22.3 region. Further
analysis of the deletion revealed monosomy of the PCNT,
DIP2A, S100B and PRMT2 genes (27). Of interest, certain SNPs of
PCNT located in this 200 kb region have been associated with
schizophrenia (28) and major
depressive disorder (29). The second
candidate gene in the etiology of major mood disorders is
S100B, encoding S100 calcium-binding protein B, which has
been associated with the etiology of schizophrenia (30), bipolar depression (31,32) and
autism (33). However, these studies
predominantly refer to protein overexpression compared with levels
observed for healthy controls. In the current case, a
downregulation of S100B would be expected due to the
haploinsufficiency of the examined region; however, further
experiments are required to support this hypothesis. Furthermore,
CNVs in the 21q22.3 region, including DIP2A and
S100B, have been associated with ASD (34). A frame shift mutation and a splice
site variant of DIP2A were observed in two families with an
ASD affected member each (35,36).
Furthermore, DIP2A has been associated with the onset of
dyslexia (37), a clinical
characteristic missing in the patient presented in the current
study, but described for patients with monosomy of the 21q22.3
region (37). DIP2A was
suggested to be a fragile X mental retardation 1 translation
regulator target in nervous cells (38).
In conclusion, the present study described the case
of a 10-year old male, where aCGH analysis identified one of the
shortest deletion/duplication of the 21q22.3 region reported to
date. This information may guide genetic counseling in the future.
Furthermore, it was hypothesized that the presence of a
susceptibility locus for ASD may be associated with depression and
anxiety; this locus was mapped in a 200 kb region between
PCNT and PRMT2.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
SO, IP, LT, EM and EP conceived and designed the
study. ES, DTP, SS, PN, ME acquired and analyzed the data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Written informed written consent was obtained from
the patients' parents and the study protocol was approved by the
Ethics Committee of the ‘P. & A. Kyriakou’ Children's Hospital
(Athens, Greece).
Patient consent for publication
The family of the patient gave a written consent for
the publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Theisen A and Shaffer LG: Disorders caused
by chromosome abnormalities. Appl Clin Genet. 3:159–174.
2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Canfield MA, Honein MA, Yuskiv N, Xing J,
Mai CT, Collins JS, Devine O, Petrini J, Ramadhani TA, Hobbs CA, et
al: National estimates and race/ethnic-specific variation of
selected birth defects in the United States, 1999-2001. Birth
Defects Res A Clin Mol Teratol. 76:747–756. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Manolakos E, Peitsidis P, Eleftheriades M,
Dedoulis E, Ziegler M, Orru S, Liehr T and Petersen MB: Prenatal
detection of full monosomy 21 in a fetus with increased nuchal
translucency: Molecular cytogenetic analysis and review of the
literature. J Obstet Gynaecol Res. 36:435–440. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Errichiello E, Novara F, Cremante A, Verri
A, Galli J, Fazzi E, Bellotti D, Losa L, Cisternino M and Zuffardi
O: Dissection of partial 21q monosomy in different phenotypes:
Clinical and molecular characterization of five cases and review of
the literature. Mol Cytogenet. 9(21)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fukai R, Hiraki Y, Nishimura G, Nakashima
M, Tsurusaki Y, Saitsu H, Matsumoto N and Miyake N: A de novo
1.4-Mb deletion at 21q22.11 in a boy with developmental delay. Am J
Med Genet A. 164A:1021–1028. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lyle R, Béna F, Gagos S, Gehrig C, Lopez
G, Schinzel A, Lespinasse J, Bottani A, Dahoun S, Taine L, et al:
Genotype-phenotype correlations in Down syndrome identified by
array CGH in 30 cases of partial trisomy and partial monosomy
chromosome 21. Eur J Hum Genet. 17:454–466. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roberson EDO, Wohler ES, Hoover-Fong JE,
Lisi E, Stevens EL, Thomas GH, Leonard J, Hamosh A and Pevsner J:
Genomic analysis of partial 21q monosomies with variable
phenotypes. Eur J Hum Genet. 19:235–238. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ilbery PL, Lee CW and Winn SM: Incomplete
trisomy in a mongoloid child exhibiting minimal stigmata. Med J
Aust. 48:182–184. 1961.PubMed/NCBI
|
|
9
|
Pelleri MC, Cicchini E, Locatelli C,
Vitale L, Caracausi M, Piovesan A, Rocca A, Poletti G, Seri M,
Strippoli P, et al: Systematic reanalysis of partial trisomy 21
cases with or without Down syndrome suggests a small region on
21q22.13 as critical to the phenotype. Hum Mol Genet. 25:2525–2538.
2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wechsler D: Manual for the Wechsler
Intelligence Scale for children. 3rd edition. The Psychological
Corporation, San Antonio, TX. 1991.
|
|
11
|
American Psychiatric Association:
Diagnostic and statistical manual of mental disorders. 5th edition.
American Psychiatric Association, Arlington, VA, 2013.
|
|
12
|
ISCN 2016: An International System for
Human Cytogenomic Nomenclature. McGowan-Jordan J, Simons A and
Schmid M (eds). Cytogenet Genome Res 149: 237-328, 2016.
|
|
13
|
Papoulidis I, Sotiriadis A, Siomou E,
Papageorgiou E, Eleftheriades M, Papadopoulos V, Oikonomidou E,
Orru S, Manolakos E and Athanasiadis A: Routine use of array
comparative genomic hybridization (aCGH) as standard approach for
prenatal diagnosis of chromosomal abnormalities. Clinical
experience of 1763 prenatal cases. Prenat Diagn. 35:1269–1277.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Bonaglia MC, Marelli S, Gottardi G, Zucca
C, Pramparo T, Giorda R, Grasso R, Borgatti R and Zuffardi O:
Subtelomeric trisomy 21q: A new benign chromosomal variant. Eur J
Med Genet. 50:54–59. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gijsbers ACJ, van Haeringen A, Bosch CAJ,
Hansson K, Verschuren M, Bakker E, Breuning MH and Ruivenkamp CAL:
A subtle familial translocation t(3;21)(p26.3;q22.3): An apparently
healthy boy with a 3p deletion and 21q duplication. Cytogenet
Genome Res. 128:245–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Su MT, Kuan LC, Chou YY, Tan SY Kuo TC and
Kuo PL: Partial trisomy of chromosome 21 without the Down syndrome
phenotype. Prenat Diagn. 36:492–495. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Ravnan JB, Tepperberg JH, Papenhausen P,
Lamb AN, Hedrick J, Eash D, Ledbetter DH and Martin CL: Subtelomere
FISH analysis of 11,688 cases: An evaluation of the frequency and
pattern of subtelomere rearrangements in individuals with
developmental disabilities. J Med Genet. 43:478–489.
2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rowe LR, Lee JY, Rector L, Kaminsky EB,
Brothman AR, Martin CL and South ST: U-type exchange is the most
frequent mechanism for inverted duplication with terminal deletion
rearrangements. J Med Genet. 46:694–702. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yu S and Graf WD: Telomere capture as a
frequent mechanism for stabilization of the terminal chromosomal
deletion associated with inverted duplication. Cytogenet Genome
Res. 129:265–274. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Falik-Borenstein TC, Pribyl TM, Pulst SM,
Van Dyke DL, Weiss L, Chu ML, Kraus J, Marshak D and Korenberg JR:
Stable ring chromosome 21: Molecular and clinical definition of the
lesion. Am J Med Genet. 42:22–28. 1992.PubMed/NCBI View Article : Google Scholar
|
|
21
|
McGinniss MJ, Kazazian HH Jr, Stetten G,
Petersen MB, Boman H, Engel E, Greenberg F, Hertz JM, Johnson A,
Laca Z, et al: Mechanisms of ring chromosome formation in 11 cases
of human ring chromosome 21. Am J Hum Genet. 50:15–28.
1992.PubMed/NCBI
|
|
22
|
Bassett AS, Chow EW and Weksberg R:
Chromosomal abnormalities and schizophrenia. Am J Med Genet.
97:45–51. 2000.PubMed/NCBI
|
|
23
|
Janowsky DS and Davis JM: Diagnosis and
treatment of depression in patients with mental retardation. Curr
Psychiatry Rep. 7:421–428. 2005.PubMed/NCBI
|
|
24
|
Strang JF, Kenworthy L, Daniolos P, Case
L, Wills MC, Martin A and Wallace GL: Depression and anxiety
symptoms in children and adolescents with autism spectrum disorders
without intellectual disability. Res Autism Spectr Disord.
6:406–412. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kaneva RP, Chorbov VM, Milanova VK, Kostov
CS, Nickolov KI, Chakarova CF, Stoyanova VS, Nikolova-Hill AN,
Krastev SK, Onchev GN, et al: Linkage analysis in bipolar pedigrees
adds support for a susceptibility locus on 21q22. Psychiatr Genet.
14:101–106. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu J, Juo SH, Terwilliger JD, Grunn A,
Tong X, Brito M, Loth JE, Kanyas K, Lerer B, Endicott J, et al: A
follow-up linkage study supports evidence for a bipolar affective
disorder locus on chromosome 21q22. Am J Med Genet. 105:189–194.
2001.PubMed/NCBI
|
|
27
|
Poelmans G, Engelen JJM, Van
Lent-Albrechts J, Smeets HJ, Schoenmakers E, Franke B, Buitelaar
JK, Wuisman-Frerker M, Erens W, Steyaert J, et al: Identification
of novel dyslexia candidate genes through the analysis of a
chromosomal deletion. Am J Med Genet B Neuropsychiatr Genet.
150B:140–147. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Numata S, Nakataki M, Iga J, Tanahashi T,
Nakadoi Y, Ohi K, Hashimoto R, Takeda M, Itakura M, Ueno S, et al:
Association study between the pericentrin (PCNT) gene and
schizophrenia. Neuromolecular Med. 12:243–247. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Numata S, Iga J, Nakataki M, Tayoshi S,
Tanahashi T, Itakura M, Ueno S and Ohmori T: Positive association
of the pericentrin (PCNT) gene with major depressive disorder in
the Japanese population. J Psychiatry Neurosci. 34:195–198.
2009.PubMed/NCBI
|
|
30
|
Aleksovska K, Leoncini E, Bonassi S,
Cesario A, Boccia S and Frustaci A: Systematic review and
meta-analysis of circulating S100B blood levels in schizophrenia.
PLoS One. 9:e106342:2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dagdan E, Morris DW, Campbell M, Hill M,
Rothermundt M, Kästner F, Hohoff C, von Eiff C, Krakowitzky P, Gill
M, et al: Functional assessment of a promoter polymorphism in
S100B, a putative risk variant for bipolar disorder. Am J Med Genet
B Neuropsychiatr Genet. 156B:691–699. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
da Rosa MI, Simon C, Grande AJ, Barichello
T, Oses JP and Quevedo J: Serum S100B in manic bipolar disorder
patients: Systematic review and meta-analysis. J Affect Disord.
206:210–215. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Guloksuz SA, Abali O, Aktas Cetin E,
Bilgic Gazioglu S, Deniz G, Yildirim A, Kawikova I, Guloksuz S and
Leckman JF: Elevated plasma concentrations of S100 calcium-binding
protein B and tumor necrosis factor alpha in children with autism
spectrum disorders. Br J Psychiatry. 39:195–200. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Egger G, Roetzer KM, Noor A, Lionel AC,
Mahmood H, Schwarzbraun T, Boright O, Mikhailov A, Marshall CR,
Windpassinger C, et al: Identification of risk genes for autism
spectrum disorder through copy number variation analysis in
Austrian families. Neurogenetics. 15:117–127. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Iossifov I, Ronemus M, Levy D, Wang Z,
Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et
al: De novo gene disruptions in children on the autistic spectrum.
Neuron. 74:285–299. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Iossifov I, Levy D, Allen J, Ye K, Ronemus
M, Lee YH, Yamrom B and Wigler M: Low load for disruptive mutations
in autism genes and their biased transmission. Proc Natl Acad Sci
USA. 112:E5600–E5607. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kong R, Shao S, Wang J, Zhang X, Guo S,
Zou L, Zhong R, Lou J, Zhou J, Zhang J, et al: Genetic variant in
DIP2A gene is associated with developmental dyslexia in Chinese
population. Am J Med Genet B Neuropsychiatr Genet. 171B:203–208.
2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Darnell JC, Van Driesche SJ, Zhang C, Hung
KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al:
FMRP stalls ribosomal translocation on mRNAs linked to synaptic
function and autism. Cell. 146:247–261. 2011.PubMed/NCBI View Article : Google Scholar
|