Introduction
Merosin deficient congenital muscular dystrophy type
1A (MDC1A) or laminin-α2 related muscular dystrophy (OMIM entry,
#607855) is one of the most common forms of congenital muscular
dystrophy (CMD), and accounts for 30% of all cases of CMD in
European countries (1). CMD may
appear during early onset or later in a patient's life, and is
characterized by proximal weakness, contractures, delayed motor
development, white matter abnormalities and spiral scoliosis
(2,3).
CMD is a hereditary disorder caused by recessive mutations in the
LAMA2 gene, which is located on chromosome 6q22-2, spans
over 260 kb and is comprised of 65 exons. LAMA2 encodes the
laminin-α2 chain which attaches with laminin-β1 and laminin-γ1
chain to form the heterotrimeric protein laminin 211. Laminin 211
is a primary component of the skeletal muscle basement membrane and
the extracellular matrix (1,4,5). The
protein interacts with other matrix macromolecules and contributes
to cell differentiation, cell movement and tissue phenotypes
(4). The majority of reported genetic
variations of MDC1A are homozygous or compound heterozygous
variants (6,7). De novo variations in contrast are
not common events and only a few have been reported in patients
with MDC1A (8-10).
It is estimated that the prevalence of MDC1A is
1-9/1,000,000 individuals, and accounts for 1-6% of all CMD cases
(1,2).
MDC1A is more common in Caucasians and rarer in the Asian
population (2). However, the
prevalence of the disease in the Asian population may be higher
than expected due to a lack of widespread availability of
appropriate diagnostic testing (11).
In the present study, the case of a Vietnamese male child
exhibiting clinical signs of muscular dystrophy is reported. The
child was previously undiagnosed due to his rare clinical
presentation and a lack of appropriate testing. Whole exome
sequencing (WES) was performed on the patient and his parents. WES
showed that the proband harbored two variants in the LAMA2
gene. Genotype-phenotype analysis suggested that the patient had
classical early onset MDC1A. Additionally, the de novo
variant was suggested to serve a contributive role in the
development of the disease. To the best of our knowledge, the
present study is the first to report a case of MDC1A in a
Vietnamese patient.
Patients and methods
Sample collection
Prior to collection of samples, written informed
consent form was obtained from the parents for research collection
and use of biological samples (blood and muscle) and clinical
information including the results of the magnetic resonance imaging
(MRI) scan, muscle biopsies and genetic testing. The present study
was approved by the Institutional Ethical Review Board of Vinmec
International General Hospital (Hanoi, Vietnam). From both the
patient and his parents, ~2 ml of peripheral blood was collected.
Blood samples were stored in EDTA containing tubes at -80˚C. The
deltoid was chosen as the muscle to obtain a biopsy from, and a
standard procedure was followed (12).
Muscle pathological analysis
To examine the morphology of the muscle, muscle
tissue sections were obtained, and were fixed and stained using
hematoxylin and eosin staining, as described previously (13). Needle electromyography (EMG) was
performed at the quadriceps and gastrocnemius muscle using a
Nicolet EDX system (Natus Medical, Inc.).
WES
Genomic DNA was extracted from peripheral blood
lymphocytes using a DNA Mini Blood Isolation kit according to the
manufacturer's protocol (Qiagen GmbH). A total of 50 ng genomic DNA
was used for library construction using a Nextera Rapid Capture kit
(Illumina, Inc.) according to the manufacturer's protocol.
Paired-end sequencing with a read length of 75x2 bp was performed
using a HiSeq 4000 (Illumina, Inc.).
Variant identification and
analysis
Burrows Wheeler Aligner (14) was used to map short reads to the human
reference genome (GRCh37). Platypus program version 0.8.1(15) and Genome Analysis Toolkit version 3.6
(16,17) were both used for variant calling.
Variants with a minor allele frequency >1%, as reported in the
1000 Genomes Project database (18),
were removed. SnpEff program version 4.3g (19) and the Human Genome Variant Database
(20) were used for variant
annotation. PolyPhen-2(21) and
PROVEAN version 1.13(22) were both
used to predict the impact of missense variant. PolyPhen-2 presents
the damaging score as a value between 0 and 1, where a score closer
to 1 indicates a high probability of the substitution being
damaging. For PROVEAN, if the score is ≤-2.5 (which is used as a
threshold), this indicates the variant is damaging. Protein
structure analysis was performed using HOPE (23).
Sanger validation
The candidate variants from WES were confirmed using
Sanger sequencing on an ABI 3500 DX system with BigDye Terminator
version 3.1 (Thermo Fisher Scientific, Inc.). The primer sequences
used for validation were: NM_000426.3:c.1964T>C forward,
5'-TGAGGGTGG AGGATACAAATATAG-3' and reverse, 5'-AGGGCTCCGTT
CTTATCTGC-3'; and NG_008678.1:c.3556-13T>A forward,
5'-AAGATTTACGTCCTGCCATGC-3' and reverse, 5'-CCA
TGTGGGCAACAATCTCTG-3'.
Results
Case presentation
The proband was a male, born by caesarean section at
36 weeks of gestation to healthy and non-consanguineous Vietnamese
parents with no family history of any inherited disease. His weight
was 3.4 kg and was considered as of no clinical concern at birth.
The boy was reported to show a delay in motor milestone acquisition
during his life. When visiting our hospital, the proband was 14
years old and he presented with gross motor developmental delay,
severe spiral scoliosis and limb-girdle muscular dystrophy. He
showed normal intelligence for his age, without any sign of
ophthalmoplegia or elongated face and could speak normally. The
creatine kinase (CK) levels were normal (126 U/l; Table I). The boy was not able to sit or
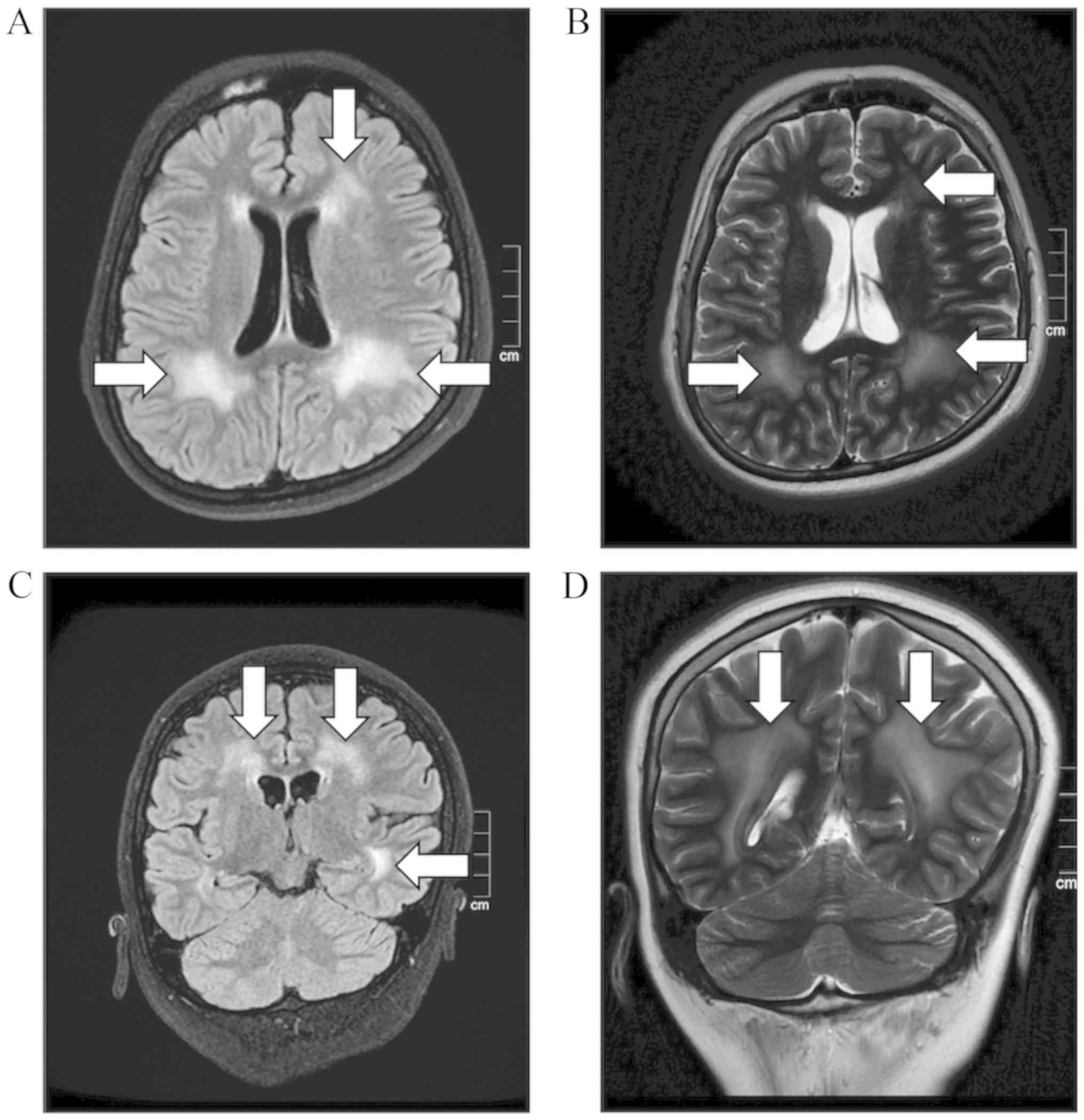
ambulate unsupported. A brain MRI scan revealed an increased signal
at the frontal and the bilateral occipital lobe. Diffuse brain
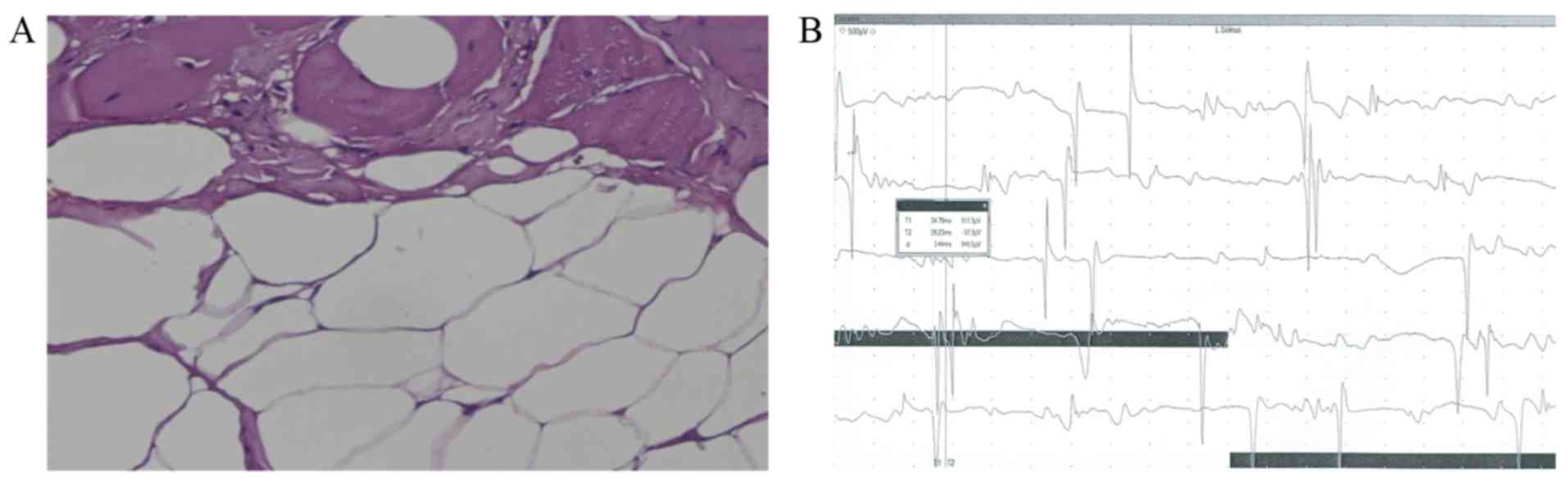
white matter hypointensity was also observed (Fig. 1). Deltoid muscle biopsy indicated
fibrous-adipose replacement (Fig.
2A). In addition, needle EMG showed the motor units exhibited
clear characteristics of myopathy (Fig.
2B) (24).
 | Table IClinical features of the patient. |
Table I
Clinical features of the patient.
| Age of onset | At birth |
| Age when
diagnosed | 14 years old |
| Sex | Male |
| Ethnic group | Kinh Vietnamese |
| Height | 146 cm |
| Weight | 44.2 kg |
| Brain MRI | WMH on T2-weighted
image |
| Mental
retardation | No |
| Independent
ambulation | No |
| Facial
dysmorphy | No |
| Motor
milestone | Sat supported |
| Contractures | Yes |
| Scoliosis | Yes |
| EMG myopathic
changes | Yes |
| Creatine
kinase | 126
U/la |
To evaluate gross motor functions, several tools,
including the Gross Motor Function Classification System (GMFCS)
(25,26), Gross Motor Function Measure 88 scale
(GMFM-88) for children with cerebral palsy aged 12-18 years old
(27), and the modified Ashworth
Scale were used to measure muscle spasticity (28). The proband was scored at level V based
on the GMFSC scale, meaning that he had severe limitations and
entirely relied on a wheelchair for movement. GMFM-88 evaluation
showed that the patient achieved 38 points in domain ‘Lying and
Rolling’, 31 points in domain ‘Sitting’, one point in domain
‘Crawling and Kneeling’, and zero points in domains ‘Standing’,
‘Walking’, ‘Running’ and ‘Jumping’. Modified Ashworth evaluations
indicated that the scores of the upper and lower limbs ranged from
3-4 points, indicating that there was a considerable increase in
muscle tone, and rigidity in flexion or extension.
Genetic studies
A total of 110, 86 and 101 million paired-end reads
were obtained from the proband, and the father and the mothers
genomes, respectively, where 95% of the reads had a Phred-score ≥30
(95% of the reads were called with a correct probability ≥99.9%)
(29). Average coverage at the
targeted regions were 81X, 66X and 80X for the proband, father and
mother, respectively. Two variants were detected in the
LAMA2 gene (Reference sequence: NM_000426.3) of the proband
and these variants were not previously described in the Vietnamese
genetic variation database (genomes.vn/) (30) indicating their rare frequency in the
population. Of these, the missense variant,
NM_000426.3:c.1964T>C, p.Leu655Pro, located in exon 14, resulted
in a substitution of leucine to proline at the 655th amino acid
residue. It appeared as a de novo variant as it was not
observed in the parents. This variant has been not listed in the
Leiden Open Variation Database
(databases.lovd.nl/shared/genes/LAMA2) or elsewhere suggesting its
novelty. In addition, WES results indicated that the proband and
his mother carried a splice site variant
(NG_008678.1:c.3556-13T>A) which was present at the intron-exon
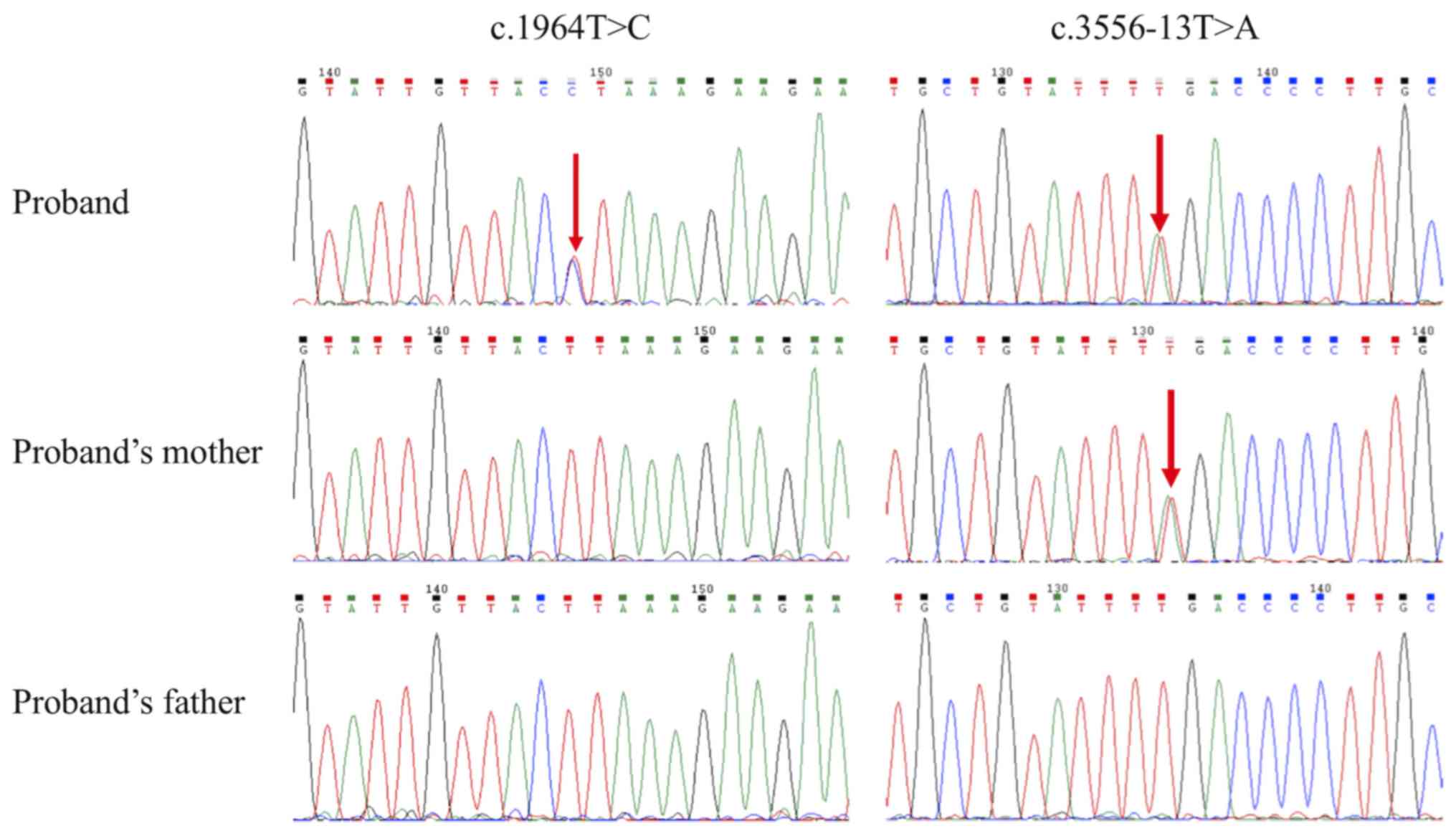
boundary of exon 25 of the LAMA2 gene. Sanger sequencing
confirmed the findings from WES where the missense variant
(NM_000426.3:c.1964T>C) was only detected in the proband;
whereas the splice site variant (NG_008678.1:c.3556-13T>A) was
found in the proband and his mother, but not in the father
(Fig. 3). The splice site variant has
been previously observed in two Chinese patients with MDC1A and is
reported to cause mRNA splicing (6).
In silico predictions
PolyPhen-2 predicted that the missense variant
(NM_000426.3:c.1964T>C) was damaging with a score of 1. PROVEAN
predicted that this variant was ‘deleterious’ with a score of
-6.992. Additionally, protein structure analysis using HOPE
suggested that the missense mutant, the size of which was smaller
compared with the wildtype, may result in a loss of amino acid
interactions. Thus, a substitution of leucine to proline may
disturb a functional domain of the laminin protein and abolish the
proteins function.
Discussion
In the present study, two variations in the
LAMA2 gene of a male proband who exhibited clinical
manifestations of CMD were identified. The missense variant
(NM_000426.3:c.1964T>C) is located in the N-terminal domain of
laminin-α2. A previous study found that half of the variants
detected in a cohort of 43 patients with MDC1A were located in the
N-terminal domain (6). Therefore,
this may suggest that this domain is either more vulnerable than
the other domains to mutations, is more sensitive to mutations or
that it exhibits a crucial function on the part of the protein. WES
analysis of the proband and his parents alongside database analyses
indicated that NM_000426.3:c.1964T>C was de novo and a
novel variant. A splice site variant (NG_008678.1:c.3556-13T>A)
in the proband and his mother was also detected. This variant and
its mRNA splicing effect (premature termination of transcription)
have been previously described in two Chinese patients with MDC1A
(6).
Together, these two variants likely caused a deficit
in laminin-α2 function, chain which is a primary component of a
trimeric basement membrane glycoprotein (1,4,5). Mutations in the LAMA2 gene result
in defective function of the laminin protein with consequential
poor muscle fiber adhesion and degeneration (31). Phenotype-genotype analysis indicated
that this proband was associated with classical early onset
LAMA2-associated muscular dystrophy. Therefore, these two variants
were submitted to the Global Variome Shared LOVD database
(databases.lovd.nl/shared/individuals/00208524).
The missense variant was searched against in
multiple databases and was not found to have been previously
reported. Therefore, the present case report may be the first
report underlining genetic variations in a Vietnamese individual
with MDC1A. In addition to the rare nature of MDC1A, a lack of
common symptoms in the child, such as increased levels of CK and
ophthalmoplegia, and the genetic heterogeneity of this disease made
diagnosis difficult. CK levels are often elevated in patients with
MDC1A during the early years of a patient's life and tend to
decrease with age (32). The proband
in the present report exhibited normal levels of CK at the age of
14. Several studies have reported normal CK levels in patients with
MDC1A (6,11,33).
Therefore, age should be taken into account when interpreting the
CK level in each patient with CMD (34). In the present study, the patient was
suspected to suffer from X-linked adrenoleukodystrophy owing to his
abnormal brain MRI scan. Genetic testing of the ABCD1 gene,
defects of which cause X-linked adrenoleukodystrophy were
performed. However, there was no evidence of mutations found and
the patient remained undiagnosed prior to WES testing (data not
shown).
MDC1A exhibits a wide spectrum of genetic and
clinical heterogeneity. Immunohistochemistry (IHC) is a first-tier
test used to determine CMD-associated diseases. Commercially
available antibodies, such as clone 5H2, clone Mer3/22B2 and clone
4H8-2 are available for performing laminin protein assays (7). However, as MDC1A is extremely rare in
Asian populations, including the Vietnamese, IHC using these
specific antibodies for laminin-α2 is often not viable. Therefore,
in addition to clinical features (phenotypes, brain MRI, EMG and
muscle morphology), molecular genetic testing, such as WES, is
highly recommended (7,35). Additionally, WES should be performed
for both the patient and their parents, as it assists in improving
our understanding of the diagnostic yield for genetically
heterogeneous disorders (36), and
may result in the identification of novel variants which contribute
to this disease (37). With the
increase in the use and availability of next generation sequencing
techniques, novel variants/mutations associated with MDC1A have
been uncovered (7). The newly
discovered variant in the present study adds to the genetic
spectrum of the disease, and also suggests the contribution of a
de novo event to the autosomal recessive inheritance of
MDC1A. Therefore, de novo events should be taken into
consideration to accurately determine the occurrence risk of this
disease in future offspring.
Acknowledgements
We are grateful to the patient's family for their
participation in and support of this study.
Funding
The present study was funded by the Vinmec Health
Care System (Hanoi, Vietnam; grant no. ISC17.03).
Availability of data and materials
Data containing information on the variants were
deposited on the Global Variome Shared LOVD database
(databases.lovd.nl/shared/individuals/00208524). The datasets used
or analyzed in the present study are available from the
corresponding author on reasonable request.
Authors' contributions
KTT prepared the experiments and wrote the
manuscript. VSL performed the bioinformatics analysis. LTN and CDV
performed the clinical diagnoses. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Durbeej M: Laminin-α2 Chain-Deficient
Congenital Muscular Dystrophy: Pathophysiology and Development of
Treatment. Curr Top Membr. 76:31–60. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Allamand V and Guicheney P:
Merosin-deficient congenital muscular dystrophy, autosomal
recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of
laminin). Eur J Hum Genet. 10:91–94. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Philpot J, Sewry C, Pennock J and Dubowitz
V: Clinical phenotype in congenital muscular dystrophy: Correlation
with expression of merosin in skeletal muscle. Neuromuscul Disord.
5:301–305. 1995.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Colognato H and Yurchenco PD: Form and
function: The laminin family of heterotrimers. Dev Dyn.
218:213–234. 2000.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Holmberg J and Durbeej M: Laminin-211 in
skeletal muscle function. Cell Adhes Migr. 7:111–121.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xiong H, Tan D, Wang S, Song S, Yang H,
Gao K, Liu A, Jiao H, Mao B, Ding J, et al: Genotype/phenotype
analysis in Chinese laminin-α2 deficient congenital muscular
dystrophy patients. Clin Genet. 87:233–243. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Oliveira J, Gruber A, Cardoso M, Taipa R,
Fineza I, Gonçalves A, Laner A, Winder TL, Schroeder J, Rath J, et
al: LAMA2 gene mutation update: Toward a more comprehensive picture
of the laminin-α2 variome and its related phenotypes. Hum Mutat.
39:1314–1337. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhou J, Tan J, Ma D, Zhang J, Cheng J, Luo
C, Liu G, Wang Y and Xu Z: Identification of Two Novel LAMA2
Mutations in a Chinese Patient with Congenital Muscular Dystrophy.
Front Genet. 9(43)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu B, Ionita-Laza I, Roos JL, Boone B,
Woodrick S, Sun Y, Levy S, Gogos JA and Karayiorgou M: De novo gene
mutations highlight patterns of genetic and neural complexity in
schizophrenia. Nat Genet. 44:1365–1369. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Yu M, Zheng Y, Jin S, Gang Q, Wang Q, Yu
P, Lv H, Zhang W, Yuan Y and Wang Z: Mutational spectrum of Chinese
LGMD patients by targeted next-generation sequencing. PLoS One.
12(e0175343)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chae JH, Lee JS, Hwang H, Kim KJ, Hwang
YS, Park JD, Cheon JE, Kim IO, Choe GY and Park SH:
Merosin-deficient congenital muscular dystrophy in Korea. Brain
Dev. 31:341–346. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dubowitz V, Oldfors A and Sewry C: Muscle
Biopsy: A Practical Approach. 4th edition. Saunders Elsevier.
2013.
|
|
13
|
Fischer AH, Jacobson KA, Rose J and Zeller
R: Hematoxylin and eosin staining of tissue and cell sections. Cold
Spring Harb Protoc. 2008(pdb.prot4986)2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rimmer A, Phan H, Mathieson I, Iqbal Z,
Twigg SRF, Wilkie AOM, McVean G and Lunter G: WGS500 Consortium:
Integrating mapping-, assembly- and haplotype-based approaches for
calling variants in clinical sequencing applications. Nat Genet.
46:912–918. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Toolkit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Auton A, Brooks LD, Durbin RM, Garrison
EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and
Abecasis GR: 1000 Genomes Project Consortium: A global reference
for human genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cingolani P, Platts A, Wang L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly
(Austin). 6:80–92. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Stenson PD, Ball EV, Mort M, Phillips AD,
Shiel JA, Thomas NS, Abeysinghe S, Krawczak M and Cooper DN: Human
Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 21:577–581.
2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Venselaar H, Te Beek TAH, Kuipers RKP,
Hekkelman ML and Vriend G: Protein structure analysis of mutations
causing inheritable diseases. An e-Science approach with life
scientist friendly interfaces. BMC Bioinformatics.
11(548)2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Paganoni S and Amato A: Electrodiagnostic
evaluation of myopathies. Phys Med Rehabil Clin N Am. 24:193–207.
2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Palisano R, Rosenbaum P, Walter S, Russell
D, Wood E and Galuppi B: Development and reliability of a system to
classify gross motor function in children with cerebral palsy. Dev
Med Child Neurol. 39:214–223. 1997.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hanna SE, Bartlett DJ, Rivard LM and
Russell DJ: Reference curves for the Gross Motor Function Measure:
Percentiles for clinical description and tracking over time among
children with cerebral palsy. Phys Ther. 88:596–607.
2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Russell DJ, Rosenbaum PL, Cadman DT,
Gowland C, Hardy S and Jarvis S: The gross motor function measure:
A means to evaluate the effects of physical therapy. Dev Med Child
Neurol. 31:341–352. 1989.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bohannon RW and Smith MB: Interrater
reliability of a modified Ashworth scale of muscle spasticity. Phys
Ther. 67:206–207. 1987.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ewing B and Green P: Base-calling of
automated sequencer traces using phred. II. Error probabilities.
Genome Res. 8:186–194. 1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Le VS, Tran KT, Bui HTP, Le HTT Nguyen CD,
Do DH, Ly HTT, Pham LTD, Dao LTM and Nguyen LT: A Vietnamese human
genetic variation database. Hum Mutat. 40:1664–1675.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hayashi YK, Tezak Z, Momoi T, Nonaka I,
Garcia CA, Hoffman EP and Arahata K: Massive muscle cell
degeneration in the early stage of merosin-deficient congenital
muscular dystrophy. Neuromuscul Disord. 11:350–359. 2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sparks SE and Escolar DM: Congenital
muscular dystrophies. In: Handbook of Clinical Neurology. Griggs RC
and Amato AA (eds). Vol 101. Elsevier. pp47–79. 2011.
|
|
33
|
Canki-Klain N, Béroud C, Clarke NF, Kovac
I, Chambert S and Guicheney P: EM.P.3.01 The adult phenotype of
congenital muscular dystrophy (MDC1A) due to mutation of LAMA2.
Neuromuscul Disord. 19(574)2009. View Article : Google Scholar
|
|
34
|
Jones KJ, Morgan G, Johnston H, Tobias V,
Ouvrier RA, Wilkinson I and North KN: The expanding phenotype of
laminin α2 chain (merosin) abnormalities: Case series and review. J
Med Genet. 38:649–657. 2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Reddy HM, Cho KA, Lek M, Estrella E,
Valkanas E, Jones MD, Mitsuhashi S, Darras BT, Amato AA, Lidov HG,
et al: The sensitivity of exome sequencing in identifying
pathogenic mutations for LGMD in the United States. J Hum Genet.
62:243–252. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Retterer K, Juusola J, Cho MT, Vitazka P,
Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J,
Monaghan KG, et al: Clinical application of whole-exome sequencing
across clinical indications. Genet Med. 18:696–704. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang Y, Muzny DM, Xia F, Niu Z, Person R,
Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al: Molecular
findings among patients referred for clinical whole-exome
sequencing. JAMA. 312:1870–1879. 2014.PubMed/NCBI View Article : Google Scholar
|