Introduction
Lynch syndrome (LS) (OMIM #120435) is one of the
most common hereditary cancer syndromes with an autosomal dominant
mode of inheritance, caused by monoallelic pathogenic variants of
one of the DNA mismatch repair (MMR) genes: MLH1 (MutL
homolog 1), MSH2 (MutS homolog 2), MSH6 (MutS homolog
6) and PMS2 (PMS1 homolog 2, mismatch repair system
component), or genomic deletion of 3' region of the EPCAM
gene by inducing hypermethylation of the MSH2 promoter.
Germline heterozygous pathogenic variant carriers have an increased
risk of developing LS-associated cancer in their adult life,
primarily colorectal and endometrial cancer and less frequently,
ovary, gastrointestinal and biliary tract cancer (1). In 1999, bi-allelic inactivation of MMR
genes was identified in LS families (2,3). The
carriers were diagnosed in their childhood with specific types of
cancer distinct from those traditionally associated with LS. In
particular, the clinical features mimicked those of
neurofibromatosis type 1 (NF1), with café-au-lait macules as the
most common manifestation. It was later termed Constitutional MMR
deficiency (CMMRD) syndrome (OMIM #276300). Overall, patients were
diagnosed with hematological malignancies, brain tumors and
LS-associated tumors in their first or second decade of life.
Homozygous or compound heterozygous bi-allelic pathogenic variants
were identified involving all four MMR genes, although the
PMS2 gene appeared to be predominantly implicated,
accounting for >50% of the cases. The remaining cases were
attributed nearly equally to MSH6 and
MLH1/MSH2 genes, respectively (4). An increasing number of CMMRD cases are
being recorded. In 2014, the European Consortium of Care for CMMRD
reported 147 patients with CMMRD described in the literature
(4) and additional cases have been
continued (5-8).
However, almost all cases were Caucasian, with only a few patients
from Pakistani families (9). The
present study describes the diagnosis of CMMRD in a young Chinese
male.
Case report
Patient
The patient was admitted to the Lanzhou General
Hospital of the Chinese People's Liberation Army in June 2018 for
clinical diagnosis and treatment. Blood samples from the patient
and his family members (parents and sister) were collected
following provision of informed consent.
Germline pathogenic variant
detection
Genomic DNA was isolated from each blood sample
using a Qiagen Blood DNA kit (Qiagen, Inc.) according to the
manufacturer's protocol. The QIAseq Human Colorectal Cancer Panel
(Qiagen, Inc.) was used with next-generation sequencing approach
(NGS). For each sample, the library was constructed from 40 ng
fragmented DNA with the ligation of the adapter and sample index
followed by PCR-based enrichment according to the manufacturer's
protocol. The sequences of the primers were not provided. The
thermocycling conditions for PCR were: Initial denaturation at
95˚C; followed by 8 cycles of 98˚C for 15 sec, 68˚C for 10 min; and
a final extension step of 5 min at 72˚C. The quality of the
purified library was verified by an Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc.) with a High Sensitivity DNA kit
(Invitrogen; Thermo Fisher Scientific, Inc.). The library of each
sample was then subjected to sequencing with MiSeq or NextSeq
according to the manufacturer's protocol. The QIAseq Targeted DNA
Panel Analysis software (CLC Genomics Workbench version 12;
qiagenbioinformatics.com/products/clc-genomics-workbench/) was used
to analyze the sequencing data and to generate variant reports. The
following genes were analyzed: APC regulator of Wnt signaling
pathway (APC; OMIM #611731; NM_000038.5); MutY DNA
glycosylase (MUTYH; OMIM #604933; NM_001128425.1);
Serine/threonine kinase 11 (STK11; OMIM #602216;
NM_000455.4); SMAD4 (OMIM #600993; NM_005359.5); bone
morphogenic protein receptor type 1A (BMPR1A; OMIM #601299;
NM_004329.2); MLH1 (OMIM #120436; NM_000249.3); MSH2
(OMIM #609309; NM_000251.2); MSH6 (OMIM #600678;
NM_000179.2); and PMS2 (OMIM #600259; NM_000535.5).
Results
The patient was a 12-year-old male who was admitted
to Lanzhou General Hospital of the Chinese People's Liberation Army
because of an anal neoplasm in combination with rectal bleeding.
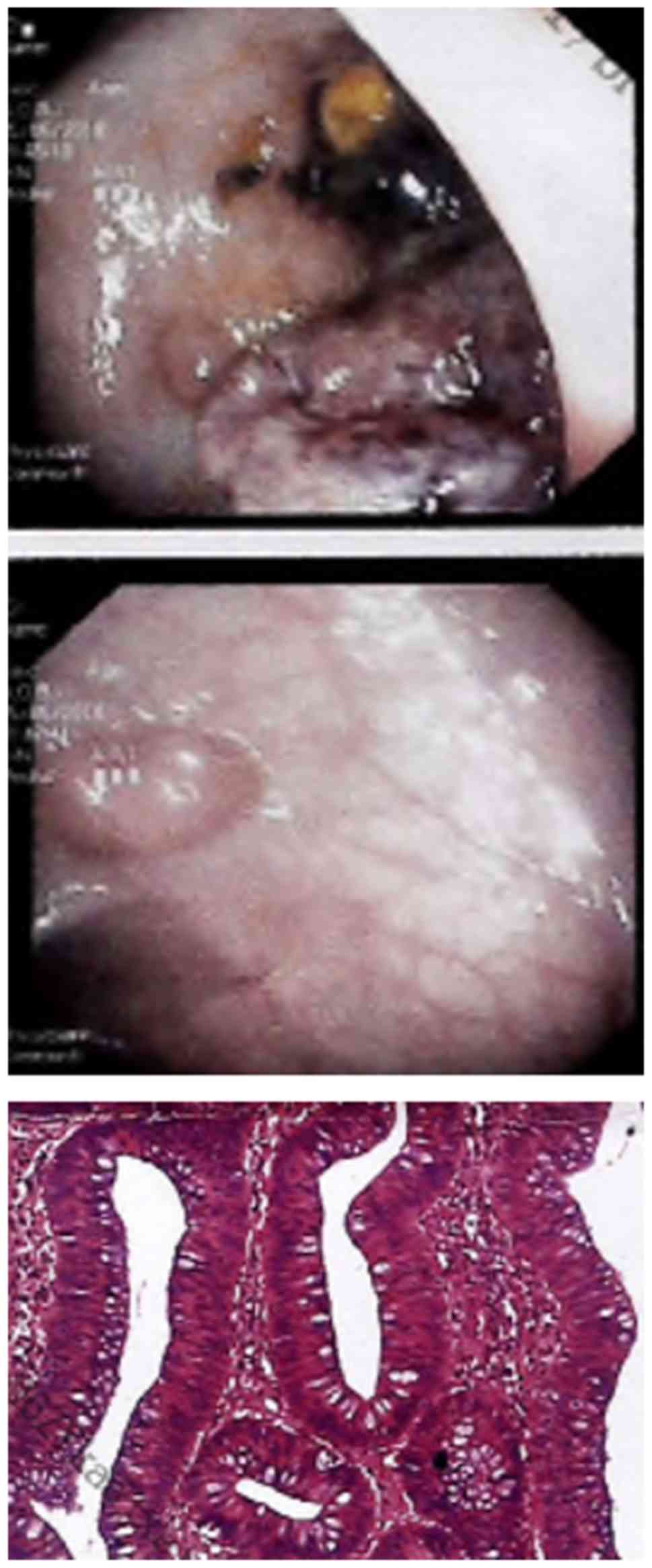
Colonoscopy examination revealed 5 polyps located at sigmoid colon
and rectum, the largest of which measured 4x4 cm in diameter
(Fig. 1). They were removed by
laparoscopic sigmoid colectomy and were identified to be tubular
adenomas, with hyperplasia/low-degree dysplasia observed in the
largest polyp. As they were not malignant tumors, neither
chemotherapy nor radiotherapy was performed. These observations led
to the diagnosis of suspected familial polyposis syndrome, in
particular the attenuated form, although the young age of the
patient did not support this diagnosis. Therefore, genetic testing
was requested, after obtaining informed consent from his parents.
An NGS approach was applied to screen an 18-gene panel including
the principal genes involved in hereditary colorectal cancer
syndromes such as APC, MUTYH, STK11,
SMAD4, BMPR1R and MMR genes MLH1, MSH2,
MSH6 and PMS2. No pathogenic variant was identified
in the APC or MUTYH genes, which are major genes
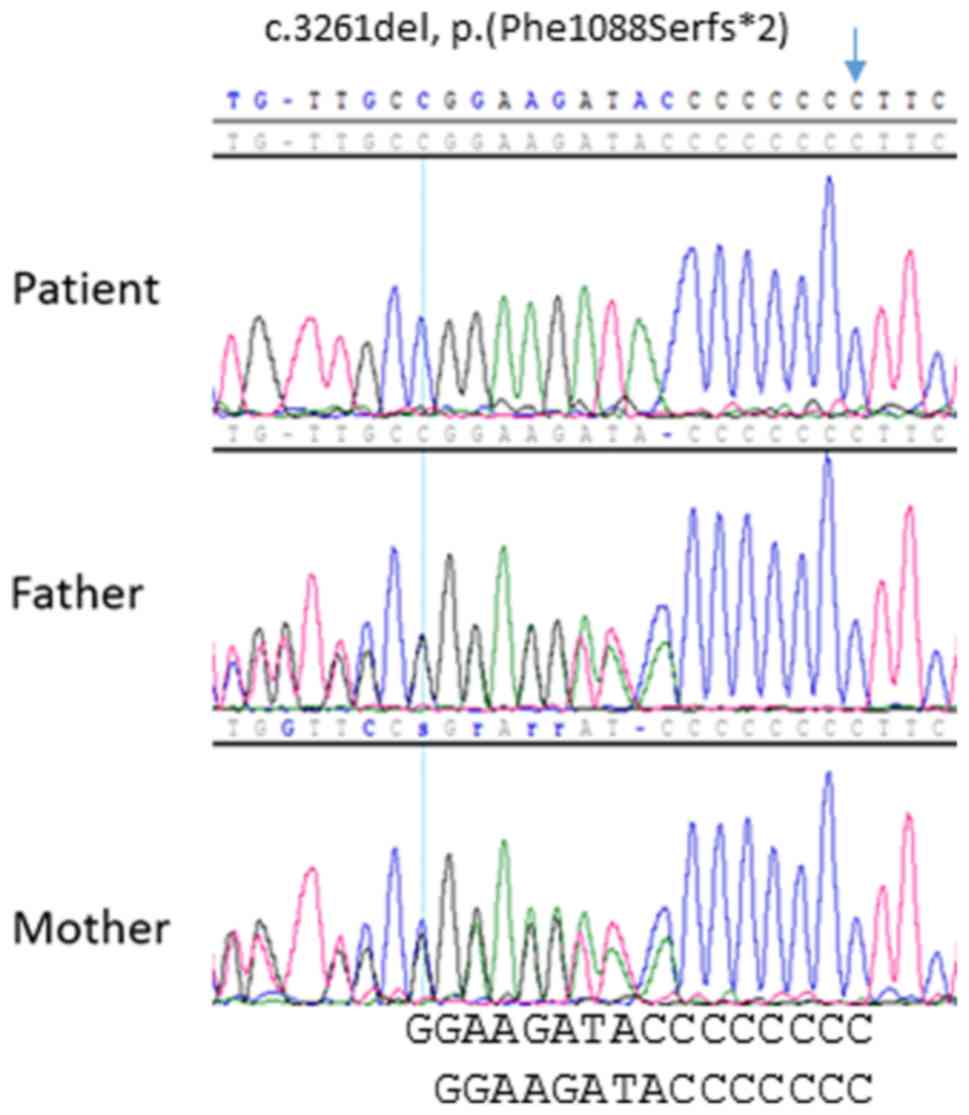
implicated in inherited colorectal polyposis (10). Instead, a pathogenic homozygous
alteration was identified in the MSH6 gene (NM_0001792):
c.3261del, p.(Phe1088Serfs*2), characterized by the
deletion of a cytosine residue in exon 5. This deleterious
frame-shift variant, leading to the interruption of protein
synthesis, has been previously identified in patients with LS,
resulting in MSH6 inactivation (insight-database.org/). The
underlying homozygous bi-allelic genotype was subsequently
confirmed through the genotype analysis of the parents, which
indicated that they were both heterozygous carriers (Fig. 2). Therefore, the diagnosis of CMMRD
was clearly established at a molecular level.
Subsequent medical examinations of the patient
revealed skin pigmentations on his back and arms, with at least one
typical ʻcafé-au-laitʼ spot in the left arm, which had appeared at
an early age. The results of an upper endoscopy did not show
gastric abnormalities. A small bowel examination was not performed,
and a magnetic resonance imaging scan of the brain indicated no
abnormalities. A bone marrow examination did not identify any
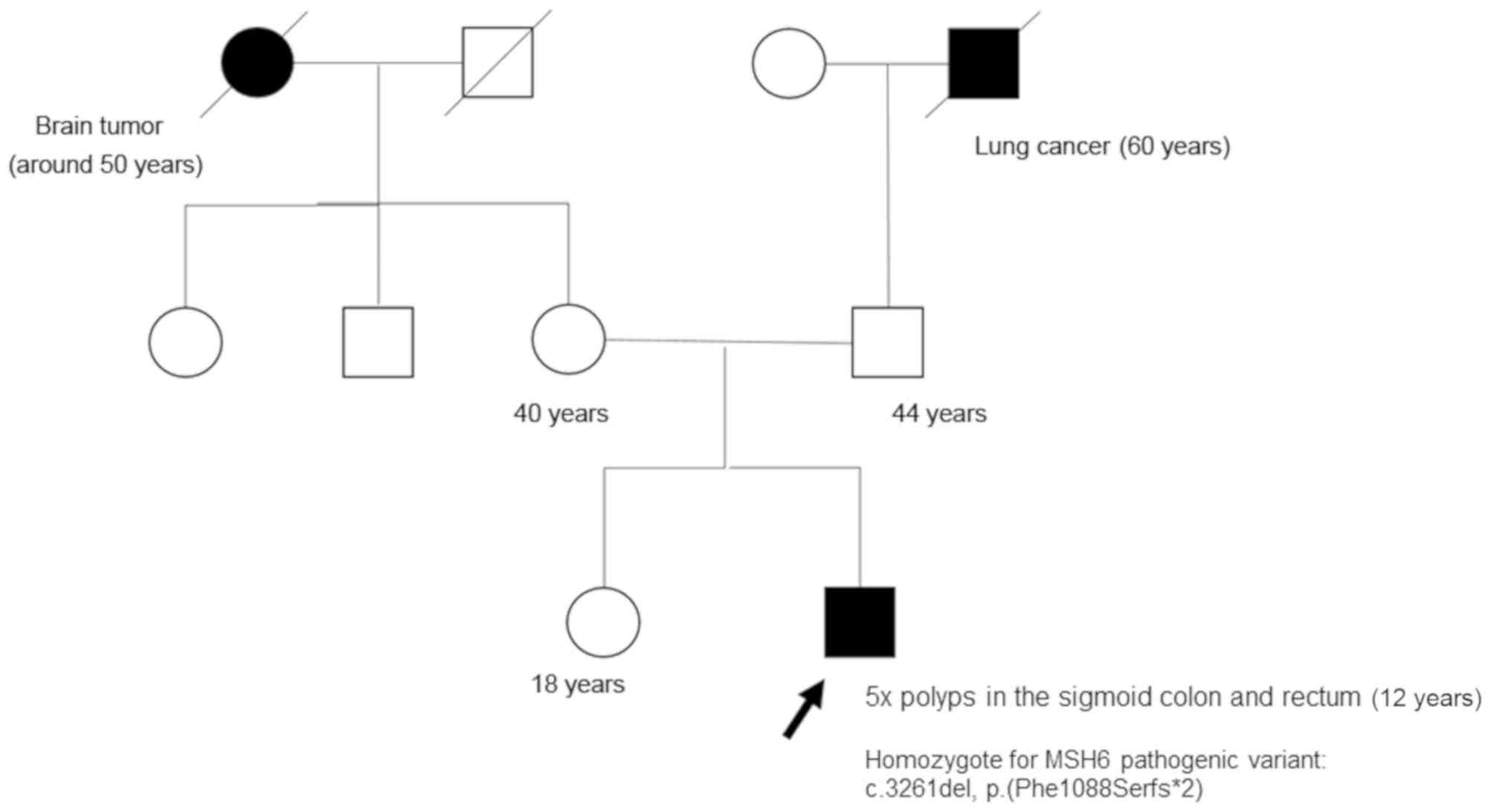
malignancy. Regarding the family (Fig.
3), the parents confirmed that there was no consanguinity in
the family. His parents, aged 44 and 40 years, respectively, and a
sister aged 18 years who was also a heterozygous carrier, were all
asymptomatic. They had all undergone recent colonoscopy
examinations, the results of which were normal. The mother had also
undergone a gynecological examination, the results of which were
normal. In the extended family, 2 members were diagnosed with
cancer: His paternal grandfather succumbed to lung cancer at the
age of 60 and his maternal grandmother succumbed to a brain tumor
in her fifties (Fig. 3). Therefore,
the origin of the homozygosity of the patient was intriguing. The
first hypothesis was that both unrelated parents carried the same
pathogenic variant by chance. However, as the variant c.3261del is
either absent or very rare and never detected in Asian population
(minor allele frequency: 0 out of 50360 alleles = 0% in East and
South Asia) (gnomadbroadinstitute.org/), the probability for a
marriage occurring between 2 unrelated carriers is quite low.
Conversely, by analyzing the NGS sequencing data, it was identified
that the patient was homozygous for all SNPs in the MSH6
locus (data not shown), despite the fact that a large number of
SNPs exists in the MSH6 locus, with high frequency of
heterozygosity in the general population. This result strongly
suggested that both parental alleles shared an identical haplotype
for this locus. This hypothesis was additionally supported by the
fact that the family had been living, for several generations, in a
mountain village with limited population. Taken together, it
appeared most likely that the frequency of the mutant allele was
highly concentrated in this village, which was the cause of the
homozygosity for the pathogenic variant without an acknowledged
consanguineous relationship.
Discussion
To the best of our knowledge, this is the first
clearly diagnosed case of CMMRD in the Chinese, or even in the
general Asian, population, indicating that CMMRD occurs regardless
of ethnic origin. The results of the present study demonstrated the
benefit of the use of a gene panel for the detection of a causative
genetic defect. Non-cancerous colorectal adenomatous polyps as the
first clinical sign has been previously reported in a number of
patients with CMMRD (9,11-14).
Notably, among 19 distinct MSH6 pathogenic variants
previously described in CMMRD (15,16), the
same pathogenic variant was identified in a case who had a similar
clinical presentation; a number of adenomatous polyps at the age of
9(13). However, the existence of a
probable genotype/phenotype correlation requires further
investigation. A previous study suggested that MSH6 is
associated with impaired antibody maturation (17); however, whether this was the case in
the patient of the present study is unknown. No immunology testing
was performed, but there was no clinical manifestation suggesting
immunodeficiency. It is well-known that LS is associated with
variable penetrance depending upon the genes involved. In small
families, it is likely that the typical phenotype may not be
exhibited. Indeed, in the family of the patient of the present
study, no clinical manifestations suggestive of LS were observed.
Therefore, we hypothesize that a number of new CMMRC cases remain
undiagnosed among pediatric patients, due to asymptomatic parents
affected by LS. In addition, the results of the present study
suggest that the risk of CMMRD may be particularly increased in
geographically isolated regions where consanguineous relationships
may occur more frequently. Genetic screening should be recommended
more systematically to young patients with hematological
malignancies, brain cancer and specific types of cancer within the
group of LS-associated malignancies, especially those who present
with café-au-lait spots, which are common features of CMMRD.
Identification of patients with CMMRD is of clinical importance for
several reasons. Firstly, for genetic counseling of the family; for
example, the parents and the sister of the family in the present
study, although asymptomatic at present, are all affected by LS as
they are heterozygous pathogenic variant carriers. Therefore,
appropriate clinical surveillance should be applied regarding the
high risk of LS-associated cancer and options for
prenatal/preimplantation diagnostics could be discussed (18). Secondly, for the clinical care of the
patient with CMMRD; it is well known that the management of the
patients diagnosed with CMMRD is challenging. Guidelines for
clinical surveillance would be helpful (4). It is worth noting that recently
developed immunotherapy (PD1 inhibition) demonstrated encouraging
responses in patients with CMMRD, further emphasizing the
importance of identifying patients with CMMRD.
Acknowledgements
The authors would like to thank Dr Zengqiang Yang,
Dr Feng Song and Dr Pengjin Sun (Lanzhou General Hospital of the
Chinese People's Liberation Army) who were involved in patient
follow-up and the collection of personal and family history. The
authors would also like to thank Dr Xiaoyue Sun, Dr Baoxia Tie, Dr
Yansheng Zhang, Dr Wei Sang (Lanzhou General Hospital of the
Chinese People's Liberation Army) for their contribution to the
case discussion and the diagnosis.
Funding
The present study was partially supported by The
National Key Research and Development Program of China (grant no.
2016YFC1302601).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MX identified the patient, collected clinical and
biological data and initiated the molecular analysis. ZY was
responsible for the patient through diagnosis and treatment. HH and
PL designed the molecular analysis approach, and conducted the
pathogenic variant screening and biological interpretation. QW
conducted the data analysis. FG coordinated and supervised the
study, was responsible for the clinical diagnosis and treatment of
the patient and the evaluation of all data. MX, ZY and QW were
involved in the writing and revision of the manuscript. HH, PL and
FG reviewed and revised the manuscript. All authors read and
approved the final mauscript.
Ethics approval and consent to
participate
Informed consent was obtained from the patients or
the legal representatives of the young patient.
Patient consent for publication
Written informed consent for publication was
obtained from the parents of the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lynch HT, Lynch JF, Lynch PM and Attard T:
Hereditary colorectal cancer syndromes: Molecular genetics, genetic
counseling, diagnosis and management. Fam Cancer. 7:27–39.
2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang Q, Lasset C, Desseigne F, Frappaz D,
Bergeron C, Navarro C, Ruano E and Puisieux A: A Neurofibromatosis
and early onset of cancers in hMLH1-deficient children. Cancer Res.
59:294–297. 1999.PubMed/NCBI
|
|
3
|
Ricciardone MD, Ozçelik T, Cevher B, Ozdağ
H, Tuncer M, Gürgey A, Uzunalimoğlu O, Cetinkaya H, Tanyeli A,
Erken E and Oztürk M: Human MLH1 deficiency predisposes to
hematological malignancy and neurofibromatosis type 1. Cancer Res.
59:290–293. 1999.PubMed/NCBI
|
|
4
|
Wimmer K, Kratz CP, Vasen HF, Caron O,
Colas C, Entz-Werle N, Gerdes AM, Goldberg Y, Ilencikova D, Muleris
M, et al: Diagnostic criteria for constitutional mismatch repair
deficiency syndrome: Suggestions of the European consortium 'care
for CMMRD' (C4CMMRD). J Med Genet. 51:355–365. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ramchander NC, Ryan NA, Crosbie EJ and
Evans DG: Homozygous germ-line mutation of the PMS2 mismatch repair
gene: A unique case report of constitutional mismatch repair
deficiency (CMMRD). BMC Med Genet. 18(40)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Suerink M, Potjer TP, Versluijs AB, Ten
Broeke SW, Tops CM, Wimmer K and Nielsen M: Constitutional mismatch
repair deficiency in a healthy child: On the spot diagnosis? Clin
Genet. 93:134–137. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tesch VK, IJspeert H, Raicht A, Rueda D,
Dominguez-Pinilla N, Allende LM, Colas C, Rosenbaum T, Ilencikova
D, Baris HN, et al: No overt clinical immunodeficiency despite
immune biological abnormalities in patients with constitutional
mismatch repair deficiency. Front Immunol. 9(1506)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Taeubner J, Wimmer K, Muleris M, Lascols
O, Colas C, Fauth C, Brozou T, Felsberg J, Riemer J, Gombert M, et
al: Diagnostic challenges in a child with early onset desmoplastic
medulloblastoma and homozygous variants in MSH2 and MSH6. Eur J Hum
Genet. 26:440–444. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
De Vos M, Hayward BE, Charlton R, Taylor
GR, Glaser AW, Picton S, Cole TR, Maher ER, McKeown CM, Mann JR, et
al: PMS2 mutations in childhood cancer. J Natl Cancer Inst.
98:358–361. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Achatz MI, Porter CC, Brugières L, Druker
H, Frebourg T, Foulkes WD, Kratz CP, Kuiper RP, Hansford JR,
Hernandez HS, et al: Cancer screening recommendations and clinical
management of inherited gastrointestinal cancer syndromes in
childhood. Clin Cancer Res. 23:e107–e114. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Trimbath JD, Petersen GM, Erdman SH, Ferre
M, Luce MC and Giardiello FM: Café-au-lait spots and early onset
colorectal neoplasia: A variant of HNPCC? Fam Cancer. 1:101–105.
2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Auclair J, Leroux D, Desseigne F, Lasset
C, Saurin JC, Joly MO, Pinson S, Xu XL, Montmain G, Ruano E, et al:
Novel biallelic mutations in MSH6 and PMS2 genes: Gene conversion
as a likely cause of PMS2 gene inactivation. Hum Mutat.
28:1084–1090. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jasperson KW, Samowitz WS and Burt RW:
Constitutional mismatch repair-deficiency syndrome presenting as
colonic adenomatous polyposis: Clues from the skin. Clin Genet.
80:394–397. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Johannesma PC, van der Klift HM, van
Grieken NC, Troost D, Te Riele H, Jacobs MA, Postma TJ, Heideman
DA, Tops CM, Wijnen JT and Menko FH: Childhood brain tumours due to
germline bi-allelic mismatch repair gene mutations. Clin Genet.
80:243–255. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ilencikova D, Sejnova D, Jindrova J and
Babal P: High-grade brain tumors in siblings with biallelic MSH6
mutations. Pediatr Blood Cancer. 57:1067–1070. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hoell JI, Gombert M, Ginzel S, Loth S,
Landgraf P, Käfer V, Streiter M, Prokop A, Weiss M, Thiele R and
Borkhardt A: Constitutional mismatch repair-deficiency and
whole-exome sequencing as the means of the rapid detection of the
causative MSH6 defect. Klin Padiatr. 226:357–361. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gardès P, Forveille M, Alyanakian MA,
Aucouturier P, Ilencikova D, Leroux D, Rahner N, Mazerolles F,
Fischer F, Kracker S and Durandy A: Human MSH6 deficiency is
associated with impaired antibody maturation. J Immunol.
188:2023–2029. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Vasen HF, Möslein G, Alonso A, Bernstein
I, Bertario L, Blanco I, Burn J, Capella G, Engel C, Frayling I, et
al: Guidelines for the clinical management of Lynch syndrome
(hereditary non-polyposis cancer). J Med Genet. 44:353–362.
2007.PubMed/NCBI View Article : Google Scholar
|