Introduction
Treacher Collins syndrome (TCS; OMIM no. 154500;
omim.org/) is a well-described autosomal dominant type
of mandibulofacial dysostosis (MFD), with an estimated prevalence
of 1 in 50,000 live births (1). The
earliest known reports of the condition came from Thomson in
1849(2) and Berry in 1889(3), but the condition is officially named
after E. Treacher Collins, who described the diagnostic criteria of
this disease in 1900(4). A more
detailed description of the syndrome was provided by Franceshetti
and Klein in 1949, who used the term MFD, and subsequently TCS was
also referred to as Franceshetti-Klein syndrome (4).
TCS is characterized by symmetrical malformations
with variable clinical features. Some typical characteristics of
the disease are down-slanting palpebral fissures, hypoplastic
zygomatic bone, lower eyelid coloboma associated with loss of the
medial eyelashes, micrognathia, ear aplasia or microtia, conductive
hearing loss and cleft palate (4).
Although the disorder is thought to be of complete penetrance,
clinical evaluation of affected patients showed incomplete
penetrance (5) There is a high degree
of intra- and interfamilial clinical heterogeneity, from severe
craniofacial malformation to patients who are so mildly affected
that they escape diagnosis (6);
however, if such patients undergo more careful examination, minor
malformations are often identified (6).
TCS exhibits genetic heterogeneity as well. To date,
three genes have been identified to be associated with this
disease: The treacle ribosome biogenesis factor 1 (TCOF1) gene on
chromosome 5q32-33.1; the RNA polymerase I and III subunit D
(POLR1D) gene on chromosome 13q12.2; and the RNA polymerase I and
III subunit C (POLR1C) gene on chromosome 6p21, whereas the
syndrome is inherited in a dominant manner through heterozygous
mutations in the TCOF1 and POLR1D genes, mutations in POLR1C gene
are inherited in an autosomal recessive manner and compound
heterozygosity is required to be phenotypically classed as having
TCS (7). TCOF1 mutations are the most
common cause of TCS, with an estimated 60% of TCS cases attributed
to de novo mutations of this gene (6,7). The TCOF1
gene encodes the treacle protein, which is a nucleolar
phosphoprotein that is trafficked between the nucleolus and
cytoplasm; it is involved in rRNA and rDNA gene transcription
affecting the proliferation and differentiation of neural crest
cells during early embryogenesis (8).
In total, >150 different mutations of TCOF1 associated with TCS,
have been reported, proposed mutational hot spots on the gene
indicate that exons 23 and 24 are responsible for >30% of
pathogenic mutations (9). In the
majority of cases, the mutations are frameshift deletions or
duplications leading to premature termination codons or mutations
in the promoter region (10,11).
The present study reports on a patient who presented
with clinical features indicative of TCS with a heterozygous
mutation in the TCOF1 gene. The mutation has not been described in
the literature before, to the best of our knowledge. Parental
clinical evaluation and testing were performed, and revealed that
the mutation was inherited. The aim of the present study was to
contribute to the understanding of the genotype-phenotype
association in patients with TCS, and to offer clinically relevant
information regarding the syndrome due to the high degree of intra-
and inter-familial phenotypic variability.
Case report
A 3-year-old boy born to an unrelated healthy mother
and a father who presented with hearing problems only, was admitted
to Achepa Hospital for Special Needs (Thessaloniki, Greece) in
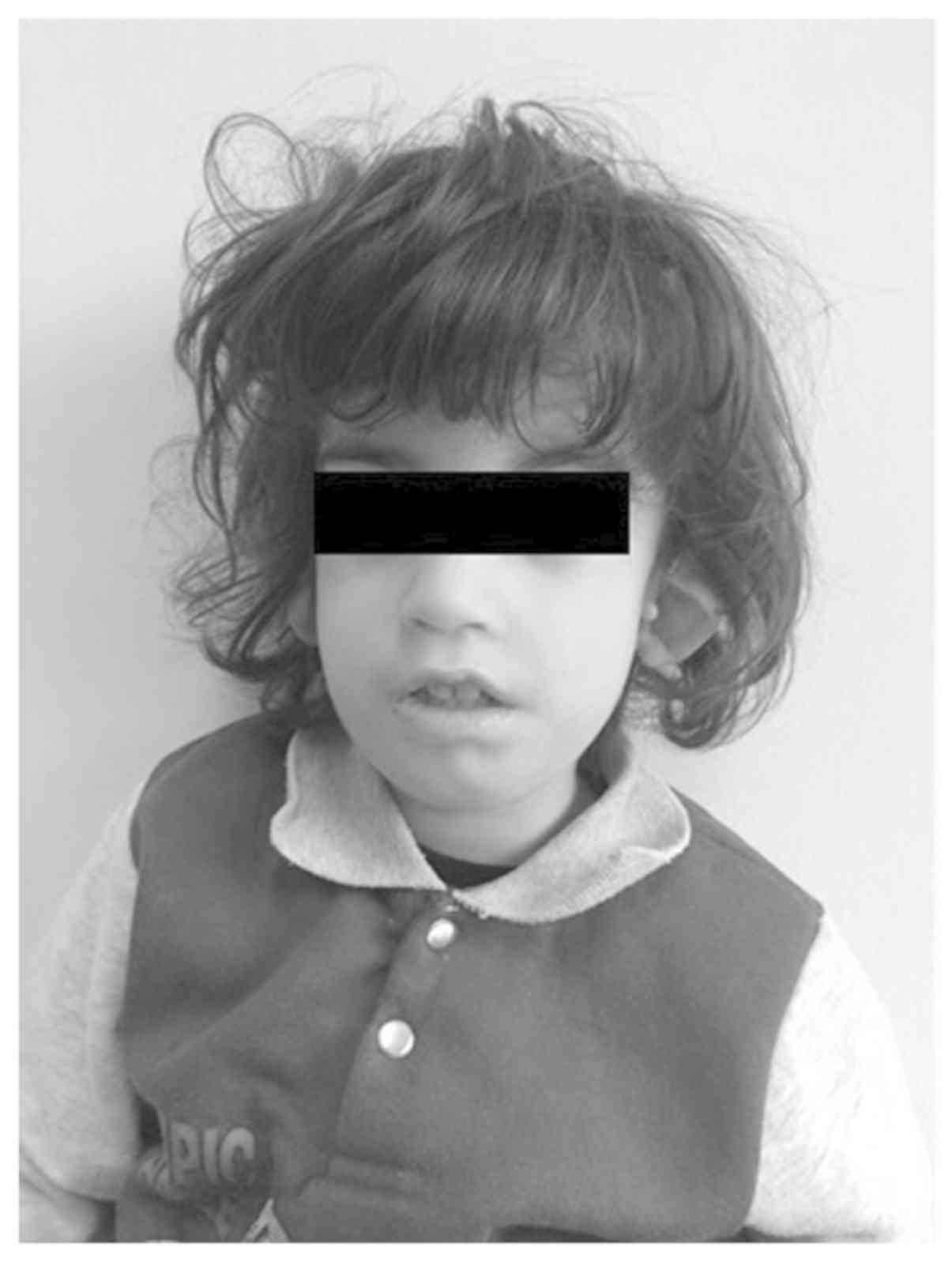
January 2017 due to phenotypic dysmorphisms. Examination of the 3
year old boy revealed external and right middle ear dysplasia,
low-set and cupped ears, almond-shaped eyes and down-slanting
palpebral fissures (Fig. 1). The
patient also had a permanent open bite and high arched palate, with
mouth breathing and tongue thrusting. Low birth weight, due to
feeding problems and micrognathia, and single transverse palmar
crease were present. The patient had normal heart function with no
heart abnormalities and exhibited mild intellectual disability
(Table I). Based on the phenotypic
features, a possible TCS diagnosis was made and genetic testing was
recommended for the proband and his parents.
 | Table IClinical features of the patient and
the father. |
Table I
Clinical features of the patient and
the father.
| Clinical features of
the patient | Yes | No | Clinical features of
patient's father | Yes | No |
|---|
| Downward-slanting
palpebral fissures | • | | Downward-slanting
palpebral fissures | | • |
| Zygomatic complex
hypoplasia | | • | Zygomatic complex
hypoplasia | | • |
| Conductive
deafness | • | | Conductive
deafness | | • |
| Mandibular
hypoplasia | • | | Mandibular
hypoplasia | | • |
| Atresia of external
ear canal | • | | Atresia of external
ear canal | | • |
| Microtia | • | | Microtia | | • |
| Lower eyelid
coloboma | • | | Lower eyelid
coloboma | | • |
| Cleft palate | • | | Cleft palate | | |
| Cardiac
malformation | | • | Cardiac
malformation | | • |
| Gastrostomy in the
neonatal period | | • | Gastrostomy in the
neonatal period | • | |
| Delayed motor
development | • | | Delayed motor
development | | • |
| Delayed speech
development | • | | Delayed speech
development | | • |
Genomic DNA from blood samples taken from the father
and the proband was extracted using the QIAamp DNA Blood Mini kit
(Qiagen, Inc.) according to the manufacturer's protocol. Regarding
the specific gene mutation in the present case, PCR was performed
for each sample using Qiagen HotStarTaq DNA polymerase was used
with the following thermocycling conditions: 95˚C for 15 min;
followed by 30 cycles of 94˚C for 30 sec, 58˚C for 60 sec and 72˚C
for 1 min; with a final extension step of 72˚C for 10 min. The
sequences of the primers were: TCOF_8 forward,
5'-TAAGGCCTCTGGACTTTATC-3' and reverse, 3'-CACAGTGAGAGGGGAGTAAG-5'.
The coding regions and the intronic flanking regions (+/-8 bp) of
the TCOF1 (OMIM no. 606847; chr5) gene were completely sequenced
(reference sequence, NM_001135243.1; ncbi.nlm.nih.gov). Sequence analysis was performed
using oligonucleotide-based target capture (QXT; Agilent
Technologies, Inc.) followed by next-generation sequencing (MiSeq;
Illumina, Inc.) using M13 sequencing primers (Thermo Fisher
Scientific, Inc.). Alignment and variant calls were generated using
the Burrows-Wheeler Aligner and Genome Analysis Toolkit version 3.6
(software.broadinstitute.org/gatk/) followed by
analysis of the predicted effects of the detected variants using
ALIGN-GVGD version 2007 (agvgd.hci.utah.edu/) and SIFT version 6.1 (sift.bii.a-star.edu.sg/). Where necessary, Sanger
sequencing was used to provide data for bases with insufficient
coverage (mean coverage <100X and/or minimum coverage of
<20X). The possibility that pseudogene sequences or
highly-homologous sequences may have interfered with the technical
ability to identify the variants present in the analysis was not be
excluded. All clinically significant and novel variants were
confirmed by independent Sanger sequencing.
Results and Discussion
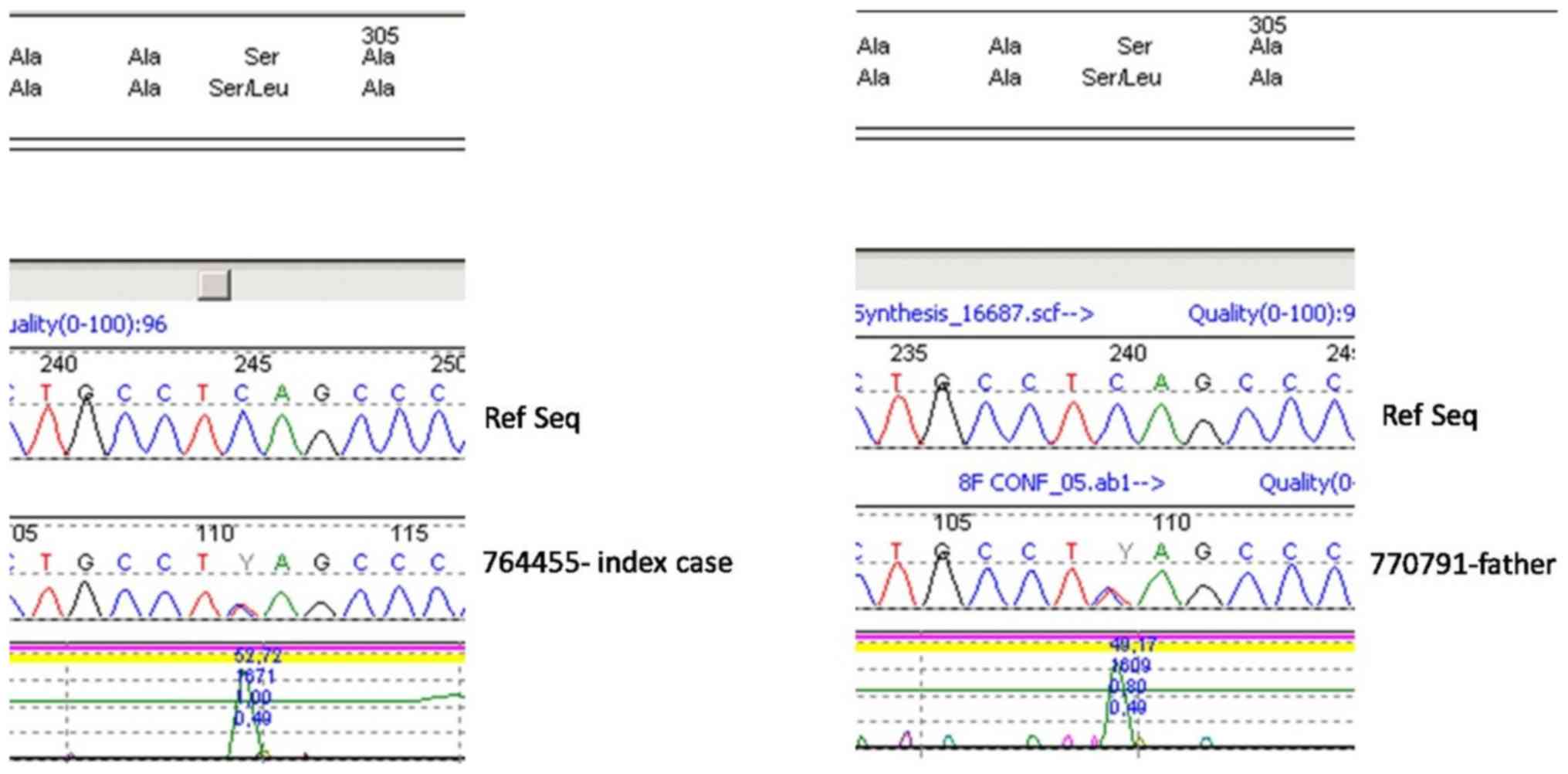
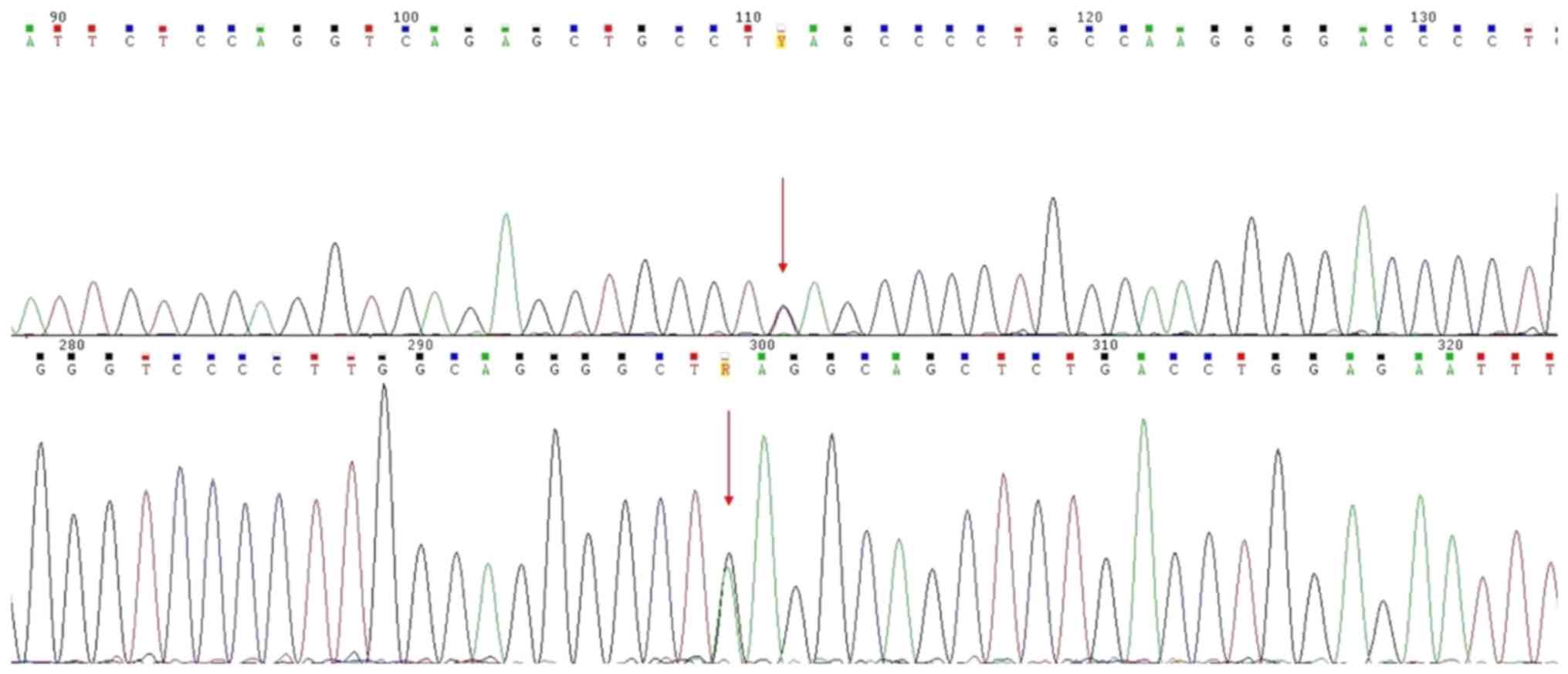
The c.911C>T (p.Ser304Leu) variant was detected
in heterozygosity in the TCOF1 gene in both the patient and his
father (Figs. 2 and 3).
In the present study, a patient with TCS and his
father, who only has hearing problems, are described. The patient
was determined to possess a previously unidentified mutation in the
TCOF1 gene which was inherited from his father. It has been
suggested that there may be hot spot mutation sites in exons 10,
15, 16, 23 and 24 of TCOF1(12), but
mutations have been identified throughout the coding region
(13). The current patient inherited
a mutation in exon 8 of TCOF1. Therefore, it is suggested that
mutational analysis of the entire TCOF1 gene should be performed if
a patient is suspected of having TCS.
There is no correlation between phenotypic
variability and the location of the mutations within TCOF1(14). In the present study the patient had a
more severe phenotype than his father, despite them both having the
same genetic variant in TCOF1 [c.911C>T (p.Ser304Leu)]. This may
be due to the association of the treacle protein with P53. Treacle,
the protein encoded for by TCOF1, is shown to have a role in
regulating transcription of rDNA and in pre-processing the rRNA
transcript (15). Inhibiting these
functions, particularly in neural crest cells, results in apoptosis
of the cells (16). However, P53
partial knockout in TCOF1+/- mice is protective against
the TCS phenotype (17).
Neural crest cell development can be divided into
three distinct stages: Formation, migration and differentiation
(18). These cells originate from
different positions along the anterior-posterior axis and they
develop into various tissues. These regions of neural crest cells
can be divided into four functional domains; cranial neural crest,
trunk neural crest, vagal and sacral neural crest, and cardiac
neural crest (19). Cranial neural
crest cells give rise to distinct cell and tissue types but are
generated transiently (19).
Therefore, it is critical that the embryo generates and maintains a
sufficient pool of neural crest progenitors that survive,
proliferate, migrate and differentiate appropriately (18). Cell lineage tracing performed in E8.5
wild-type and TCOF1+/- mouse embryos revealed that there
was no migratory nor pathfinding defects in cranial neural crest
cell migration (20). This
observation therefore indicated that TCOF1 does not play a role in
neural crest cell migration and, furthermore, that aberrant neural
crest cell migration is not the underlying cause of TCS. However,
25% fewer migrating neural crest cells were reproducibly observed
in TCS embryos compared to their wild-type mice littermates, in a
previous study (19). The deficiency
of neural crest cells arises due to extensive neuroepithelial
apoptosis from E8.0 to 10.5, which diminishes the neural stem cell
pool from which neural crest cells are derived. Therefore, TCOF1
serves a critical role in neural crest cell formation and is
required for neuroepithelial survival and neural crest cell
proliferation (20).
The primary aim of the present study was to
demonstrate the clinical heterogeneity of TCS syndrome and provide
phenotypic evidence of affected humans. The c.911C>T
(p.Ser304Leu) variant, detected in heterozygosity in the TCOF1
gene, has not been previously described in the literature, to the
best of our knowledge. This variant however is reported in dbSNP
(rs144193760; minor allele frequency, N/A; ncbi.nlm.nih.gov/snp/), ESP (minor allele frequency,
0.03%; sp.gs.washington.edu/drupal/) and gnomAD (minor allele
frequency, 0.018%, 50 heterozygous individuals reported; gnomad.broadinstitute.org/); it affects a weakly
conserved nucleotide [phyloP:0.44(-20.0;10.0)] and the predicted
effects were inconclusive regarding its pathogenicity; C15 (GV,
73.35-GD, 97.78); deleterious (score, 0.04; median, 3.36). Thus,
this mutation should be considered a variant of unknown clinical
significance. Additionally, when the syndrome is caused by TCOF1
mutations, it is inherited in an autosomal dominant manner and
significant inter- and intrafamilial clinical variability is common
in TCS. As the present analysis does not exclude variants outside
of the analyzed regions or variants which were not detected by the
methodology used, the present study states that the current result
does not confirm, but also does not exclude, that the clinical
diagnosis of TCS was caused by mutation of the TCOF1 gene, and a
more extensive analysis of the patients genome would be required to
support or discredit the gene and the specific mutations
involvement.
In conclusion, a patient with TCS and an unreported
mutation within the TCOF1 gene [c.911C>T (p.Ser304Leu)] is
described. This mutant gene was inherited from the father who only
had hearing problems. The intrafamilial phenotypic variability,
which was also observed in this case study, supports the hypothesis
that genetic modifying factors may serve a regulatory role in the
development of this syndrome.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ElP was the major contributor in writing the
manuscript. ElP, IP and EM analyzed the blood samples and found the
familial mutation. AZ and EvP analyzed the data, performed the
bibliographic research and drafted the manuscript. SF performed the
clinical examination of the patient and his family. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Informed consent for participation was obtained from
the parents.
Patient consent for publication
Informed consent for publication was obtained from
the parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Caluseriu O, Lowry BR, McLeod R, Lamont R,
Parboosingh JS, Bernier FP and Innes AM: The hutterite variant of
treacher collins syndrome: A 28-year-old story solved. Am J Med
Genet. 161A:2855–2859. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Thomson A: Notice of several cases of
malformation of the external ear, together with experiments on the
state of hearing in such persons. Mon J Med Sci. 1:420–425.
1846.
|
|
3
|
Berry GA: Note on a congenital defect
(?coloboma) of the lower lid. R Lond Ophthal Hosp Rep. 12:255–257.
1889.
|
|
4
|
Dixon MJ: Treacher collins syndrome. Hum
Mol Genet. 5:1391–1393. 1996.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dixon MJ, Marres HA, Edwards SJ, Dixon J
and Cremers CW: Treacher Collins syndrome: Correlation between
clinical and genetic linkage studies. Clin Dysmorphol. 3:96–103.
1994.PubMed/NCBI
|
|
6
|
Dixon MJ, Read AP, Donnai D, Colley A,
Dixon J and Williamson R: The gene for Treacher Collins syndrome
maps to the long arm of chromosome 5. Am J Hum Genet. 49:17–22.
1991.PubMed/NCBI
|
|
7
|
Katsanis SH and Jabs EW: Treacher Collins
Syndrome. 2004 Jul 20 (updated 2012 Aug 30). Pagon RA, Adam MP,
Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Ledbetter N,
Mefford HC, Smith RJ and Stephens K (eds): GeneReviews© (Internet).
University of Washington, Seattle, WA, 1993.2017.
|
|
8
|
Gonzales B, Henning D, So RB, Dixon J,
Dixon MJ and Valdez BC: The Treacher Collins syndrome (TCOF1) gene
product is involved in pre-rRNA methylation. Hum Mol Genet.
14:2035–2043. 2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Splendore A, Jabs EW, Félix TM and
Passos-Bueno MR: Parental origin of mutations in sporadic cases of
Treacher Collins syndrome. Eur J Hum Genet. 11:718–722.
2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Splendore A, Fanganiello RD, Masotti C,
Morganti LS and Passos-Bueno MR: TCOF1 mutation database: Novel
mutation in the alternatively spliced exon 6A and update in
mutation nomenclature. Hum Mutat. 25:429–434. 2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Masotti C, Armelin-Correa LM, Splendore A,
Lin CJ, Barbosa A, Sogayar MC and Passos-Bueno MR: A functional SNP
in the promoter region of TCOF1 is associated with reduced gene
expression and YY1 DNA-protein interaction. Gene. 359:44–52.
2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Splendore A, Silva EO, Alonso LG,
Richieri-Costa A, Alonso N, Rosa A, Carakushanky G, Cavalcanti DP,
Brunoni D and Passos-Bueno MR: High mutation detection rate in
TCOF1 among Treacher Collins syndrome patients reveals clustering
of mutations and 16 novel pathogenic changes. Hum Mutat.
16:315–322. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wise CA, Chiang LC, Paznekas WA, Sharma M,
Musy MM, Ashley JA, Lovett M and Jabs EW: TCOF1 gene encodes a
putative nucleolar phosphoprotein that exhibits mutations in
Treacher Collins syndrome throughout its coding region. Proc Natl
Acad Sci USA. 94:3110–3115. 1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Teber OA, Gillessen-Kaesbach G, Fischer S,
Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E,
Hagedorn-Greiwe M, Hammans C, et al: Genotyping in 46 patients with
tentative diagnosis of Treacher Collins syndrome revealed
unexpected phenotypic variation. Eur J Hum Genet. 12:879–890.
2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hayano T, Yanagida M, Yamauchi Y, Shinkawa
T, Isobe T and Takahashi N: Proteomic analysis of human
Nop56p-associated pre-ribosomal ribonucleoprotein complexes.
Possible link between Nop56p and the nucleolar protein treacle
responsible for Treacher Collins syndrome. J Biol Chem.
278:34309–34319. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Trainor PA, Dixon J and Dixon MJ: Treacher
Collins syndrome: Etiology, pathogenesis and prevention. Eur J Hum
Genet. 17:275–283. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jones NC, Lynn ML, Gaudenz K, Sakai D,
Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al:
Prevention of the neurocristopathy Treacher Collins syndrome
through inhibition of p53 function. Nat Med. 14:125–133.
2008.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Trainor PA: Craniofacial birth defects:
The role of neural crest cells in the etiology and pathogenesis of
Treacher Collins syndrome and the potential for prevention. Am J
Med Genet A. 152A:2984–2994. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huang X and Saint-Jeannet JP: Induction of
the neural crest and the opportunities of life on the edge. Dev
Biol. 275:1–11. 2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Dixon J, Jones NC, Sandell LL, Jayasinghe
SM, Crane J, Rey JP, Dixon MJ and Trainor PA: Tcof1/Treacle is
required for neural crest cell formation and proliferation
deficiencies that cause craniofacial abnormalities. Proc Natl Acad
Sci USA. 103:13403–13408. 2006.PubMed/NCBI View Article : Google Scholar
|