Introduction
Paroxysmal kinesigenic dyskinesia (PKD) is a rare
condition characterized by recurrent brief episodes of dystonia,
chorea, athetosis or a combination thereof, without alteration of
consciousness. PKD may be considered as a pure form, where patients
only present with dystonia, chorea and athetosis, or as a
complicated form, were patients present with PKD combined with
benign familial infantile convulsions or hemiplegic migraine
(1). Episodes are usually triggered
by sudden voluntary movements (1).
The majority of PKD cases are inherited with an autosomal dominant
pattern with complete or incomplete penetrance; although there are
reported sporadic (1). In 2011, Chen
et al (2) first reported
mutations in the gene encoding proline-rich transmembrane
protein 2 (PRRT2, chromosome 16p11.2) as the genetic
cause of PKD in eight families (2).
At present, mutations of the PRRT2 gene are the major cause
of PKD, with a frequency ranging from 40% to >90%, depending on
case ascertainment (1,3). The frameshift mutation c.649dupC
(p.Arg217Profs*8), which results in the presence of a
premature stop codon, is a hotspot mutation (4). In the present report, two cases of
sporadic PKD with sequencing analysis of the PRRT2 gene are
described, identifying a potentially novel heterozygous missense
mutation, c.955G>T (p.Val319Leu), in exon 3 of the PRR2
gene.
Case reports
Clinical presentation
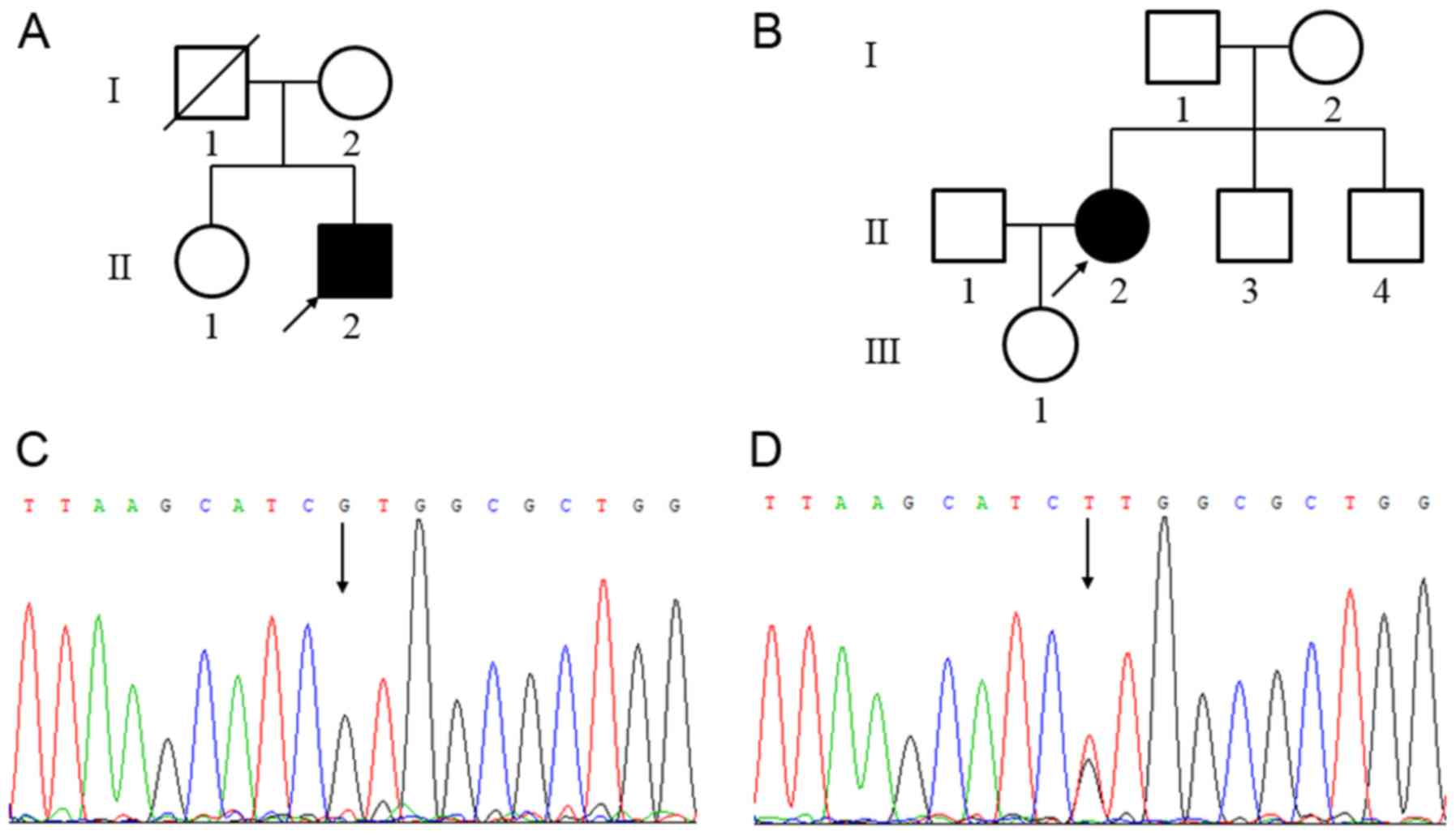
Case 1: The index patient, II:2, in Family A
(Fig. 1A), an 18-year-old male, was
referred to the neurology clinic (The Fourth Affiliated Hospital,
Zhejiang University School of Medicine), due to a 3-year history of
paroxysmal left limb dystonia. The episodes were triggered by
suddenly rising from sitting, and occasionally, being startled, and
were characterized by brief episodes of the left limbs rolling
outward involuntarily, with the wrists and ankles bent transiently,
head turning and slightly impaired speech with preserved
consciousness. The episodes usually lasted 5-10 sec with a
frequency of up to 10-20 episodes per a day. Epilepsy was suspected
initially, and 200 mg per day sodium valproate and 100 mg per day
Dilantin were prescribed. The number of episodes were reduced, but
continued to occur 3-5 times daily. Neurological examination did
not show any abnormalities. The patient had no adverse perinatal
events and had developed normally. The 24 h video
electroencephalography (VEEG) and brain magnetic resonance imaging
(MRI) were normal, and laboratory examinations, including complete
cell count, liver function tests, renal function tests, blood sugar
measurement and thyroid function were normal. PKD was diagnosed
based on the diagnostic criteria (5)
and 100 mg carbamazepine daily was prescribed. The frequency of
episodes disappeared. The patient's parents (I:1 and I:2) and older
sister (II:1) (Fig. 1A) did not
exhibit any seizures, movement disorders or migraines. The
patient's father had died in a car accident when he was 54 years
old and there was no DNA available for genetic testing.
Case 2: The index patient, II:2, in Family B
(Fig. 1B), a 31-year-old female,
presented with paroxysmal abnormal movement since the age of 12
years. She experienced dystonia and choreoathetotic movements of
the right arm; occasional clawing of the right hand; and twisted
posture, grimace and impaired speech. Each episode lasted several
seconds, and never exceeded 30 sec, without altered consciousness.
The episodes occurred ≤20 times a day. Sudden movements and
acceleration were prominent triggers. Aura-like symptom of visual
color enrichment usually preceded the episodes. The patient claimed
that walking slowly could partially control the episodes. No
positive neurological signs were identified. The results of
interictal neurological examination, brain MRI and 24 h VEEG were
normal. The patient had been prescribed 200 mg carbamazepine daily
since the age of 14 years, and a good clinical response was
achieved, and was kept on 200 mg carbamazepine every 4-5 days
since. The patient's parents (I:1 and I:2) were not consanguineous
and the patient had two healthy siblings (II:3, 29 years old and
II:4, 26 years old) and a 3-year-old child (III:1). No neurological
disorders were recorded in any other family members.
The clinical features of the two cases are
summarized in Table I.
 | Table IClinical characteristics of the two
paroxysmal kinesigenic dyskinesia cases. |
Table I
Clinical characteristics of the two
paroxysmal kinesigenic dyskinesia cases.
| Clinicopathological
characteristics | Case 1 | Case 2 |
|---|
| Sex | Male | Female |
| Family history | None | None |
| Age of onset,
years | 15 | 13 |
| Symptoms
presented | Dystonia | Dystonia,
choreoathetosis |
| Duration of
symptoms | 5-10 sec | <30 sec |
| Affected limbs | Left | Right |
| Facial
involvement | Yes | Yes |
| Presence of aura | No | Yes |
| Triggers | Sudden movements,
being startled | Sudden movements,
acceleration |
| Frequency of
episodes | 10-20 times per
day | 20-30 times per day
before 20 years old; 1-2 times per day after 20 years old |
| Combined neurological
problem | No | No |
| Response to
treatment | Symptom-free | Symptom-free |
Genetics analysis
The ethical committee of the Fourth Affiliated
Hospital, Zhejiang University School of Medicine approved the
genetics analysis. Peripheral blood samples were collected from the
patients, and their parents and siblings (Family A: I:2, II:1 and
II:2; Family B: I:1, I:2, II:2, II:3 and II:4) after obtaining
written informed consent from all the patients or the legal
representatives of the family members. Genomic DNA was extracted
using a Blood Genomic DNA Extraction kit (Qiagen, Inc.). The
primers flanking all four exons and the intron-exon boundaries of
PRRT2 (GenBank accession no. NM_145239) were designed using the
web-based Primer 3.0 program (bioinfo.ut.ee/primer3-0.4.0/) (6-8). The
PRRT2 gene was amplified by PCR. The sequences of the
primers were: Exon 2-1 forward, 5'-CCCAAGCCTATCTCC TCCTC-3' and
reverse, 5'-CTGGGTAGGGAGCTCTGG TT-3'; exon 2-2 forward,
5'-GACCCATGCCAAGAAACA GT-3' and reverse,
5'-GGATCCATGCAGAGAGGAGA-3'; exon 3 forward,
5'-TTCTGGGCTGGCTTCTCCT-3' and reverse, 5'-AAAGCTGCCCTTGCCAAC-3';
and exon 4 forward, 5'-CCCTGCTCTCTCCTGTCTGT-3' and reverse
5'-CTGTAAACAAGGCCGCTCAG-3'. Each PCR reaction consisted of 2 µl 10X
Standard Taq Reaction Buffer (Takara Bio, Inc.), 0.4 µl 10 mM
dNTPs, 0.4 µl 10 µM forward/reverse primers, 100 ng genomic DNA,
0.1 µl Taq DNA Polymerase (Takara Bio, Inc.) and 15.7 µl
nuclease-free water. The reactions were performed in thermocyclers
(PerkinElmer, Inc.), starting with an initial denaturation of 3 min
at 94˚C; followed by 30 cycles of 30 sec denaturation at 94˚C, 30
sec annealing at the primer-specific temperature, and 30 sec
extension at 72˚C. PRRT2 gene mutations were screened by
direct sequencing using an ABI Prism 3100 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The sample sequences
were compared with the genomic DNA sequence of PRRT2
(GenBank accession no. NM_145239).
An identical heterozygous missense mutation in both
patients: c.955G>T (p.Val319Leu) in exon 3 of PRRT2 was
detected (Fig. 1C and D). Screening of PRRT2 mutations in
the patients' parents and siblings did not identify any variants.
This mutation was not reported in the 1000 Genomes Project
(9), and was absent from 1,200 normal
controls (performed by Sanger sequencing), whose samples were
collected from the Medical Examination Center, The Fourth
Affiliated Hospital, Zhejiang University School of Medicine, after
providing informed consent. The 1,200 normal controls consisted of
651 men and 549 women with a median age of 45 years (range, 19-78
years). The SIFT web server (10)
prediction score of the substitution was 0.024 suggesting a
damaging effect of the substitution, and the PolyPhen-2 website
(11) prediction score was 0.997
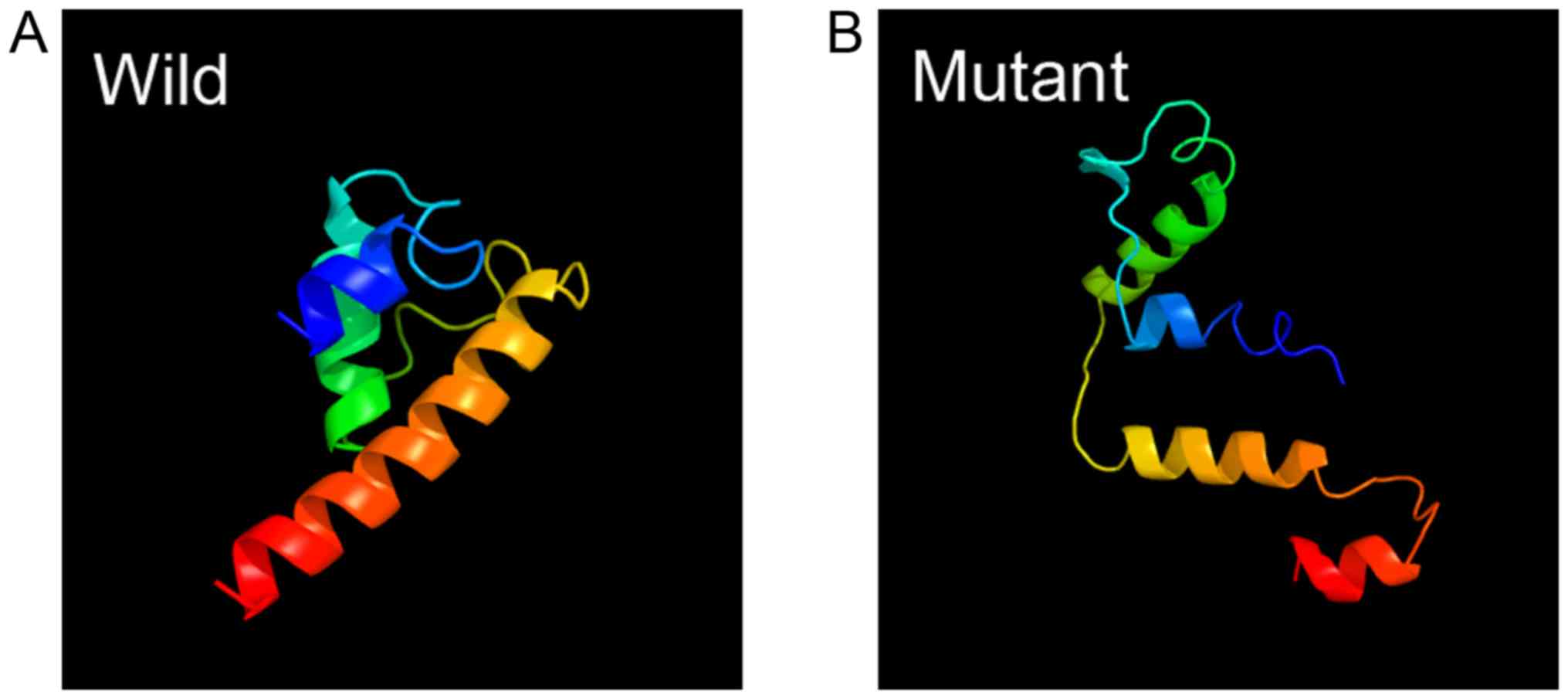
suggesting a probable damaging effect. The protein structures of
the wild-type and mutant PRRT2 were predicted using Phyre2(12). The results suggest that the structure
of mutant PRRT2 differs notably from the wild-type structure
(Fig. 2).
Discussion
PKD is often familial with autosomal dominant
inheritance, but sporadic cases of PKD have also been reported
(4,13-15),
which are attributed to incomplete penetrance or de novo
mutations (16). In the present case
report, two Chinese patients with sporadic PKD were examined and a
novel heterozygous missense mutation in exon 3 of the PRRT2
gene (c.955G>T) was identified. The family member II:1 and II:2
in Family B declined to have their 3 year old daughter (III:1)
undergo genetic analysis. The absence of the mutation in their
family members indicates the de novo status of the mutation.
This newly found variant further reinforces the importance of the
PRRT2 gene in PKD.
Compared with the commonly reported clinical
manifestation of PRRT2-associated PKD, the patients in the
present report had several primary distinctive features: i)
Sporadic occurrence; ii) onset during the teenage years; iii)
presented with pure PKD; iv) typically transitory spells, lasting
3-10 sec (always <30 sec); and v) episodes involved the limbs
and face unilaterally. Huang et al (17) summarized the clinical manifestations
and PRRT2 mutations of 110 patients with PKD, and showed
that PRRT2 mutation carriers were younger at onset and
experienced longer episodes compared with non-PRRT2 mutation
carriers (17). Previous studies have
also indicated that the majority of PRRT2 mutation-positive
patients experience bilateral episodes, and tend to present with
complicated PKD (5). The limited
sample size in the present report is inadequate for supporting the
premise that the phenotype and genotype identified are correlated.
To date, the role of PRRT2 is incompletely understood.
Previous studies have suggested that the varied mutations observed
in the PRRT2 gene may be involved in different pathogenic
molecular pathways (4,10). Thus, the function of PRRT2 and its
role in PKD requires further investigation.
In summary, a novel heterozygous missense mutation,
c.955G>T (p.Val319Leu) in exon 3 of the PRRT2 gene, was
discovered in two cases of sporadic PKD. This finding expands upon
the known mutation spectrum of the PRRT2 gene and may
provide an opportunity for further study of the genetic
pathogenesis of PKD.
Acknowledgements
Not applicable.
Funding
This work was supported by the fund of Zhejiang
Province Medical Science and Technology Project (grant nos.
2017174708 and 2020RC061) and the Natural Scientific Foundation of
Zhejiang Province (grant no. LGF20H090011).
Availability of data and materials
The datasets used or analyzed in the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
JF, SW, GZ and LC prepared the experiments and wrote
the manuscript. GZ performed the genetics analysis. JF, GZ and LC
performed the clinical diagnoses. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent from all the patients or
the legal representatives of the family members was obtained.
Informed consent was also obtained from all 1,200 healthy controls.
The Ethics Committee of the Fourth Affiliated Hospital, Zhejiang
University School of Medicine approved the genetics analysis.
Patient consent for publication
Written informed consent for publication was
obtained from the family members.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Erro R and Bhatia KP: Unravelling of the
paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry. 90:227–234.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan
GH, Guo SL, He J, Chen YF, Zhang QJ, et al: Exome sequencing
identifies truncating mutations in PRRT2 that cause paroxysmal
kinesigenic dyskinesia. Nat Genet. 43:1252–1255. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Hedera P, Xiao J, Puschmann A, Momčilović
D, Wu SW and LeDoux MS: Novel PRRT2 mutation in an African-American
family with paroxysmal kinesigenic dyskinesia. BMC Neurol.
12(93)2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhao G, Liu X, Zhang Q and Wang K: PRRT2
mutations in a cohort of Chinese families with paroxysmal
kinesigenic dyskinesia and genotype-phenotype correlation
reanalysis in literatures. Int J Neurosci. 128:751–760.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ebrahimi-Fakhari D, Saffari A,
Westenberger A and Klein C: The evolving spectrum of
PRRT2-associated paroxysmal diseases. Brain. 138:3476–3495.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Koressaar T and Remm M: Enhancements and
modifications of primer design program Primer3. Bioinformatics.
23:1289–1291. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Untergasser A, Cutcutache I, Koressaar T,
Ye J, Faircloth BC, Remm M and Rozen SG: Primer3 - new capabilities
and interfaces. Nucleic Acids Res. 40(e115)2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kõressaar T, Lepamets M, Kaplinski L,
Raime K, Andreson R and Remm M: Primer3_masker: Integrating masking
of template sequence with primer design software. Bioinformatics.
34:1937–1938. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA, et al: A global reference for human genetic
variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40 (Web Server
issue):W452–457. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kelley LA, Mezulis S, Yates CM, Wass MN
and Sternberg MJ: The Phyre2 web portal for protein modeling,
prediction and analysis. Nat Protoc. 10:845–858. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Scheffer IE, Grinton BE, Heron SE, Kivity
S, Afawi Z, Iona X, Goldberg-Stern H, Kinali M, Andrews I, Guerrini
R, et al: PRRT2 phenotypic spectrum includes sporadic and
fever-related infantile seizures. Neurology. 79:2104–2108.
2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Groffen AJ, Klapwijk T, van Rootselaar AF,
Groen JL and Tijssen MA: Genetic and phenotypic heterogeneity in
sporadic and familial forms of paroxysmal dyskinesia. J Neurol.
260:93–99. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shi CH, Sun SL, Wang JL, Liu AQ, Miao W,
Avinash C, Mao X, Tang BS and Xu YM: PRRT2 gene mutations in
familial and sporadic paroxysmal kinesigenic dyskinesia cases. Mov
Disord. 28:1313–1314. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu XR, Wu M, He N, Meng H, Wen L, Wang
JL, Zhang MP, Li WB, Mao X, Qin JM, et al: Novel PRRT2 mutations in
paroxysmal dyskinesia patients with variant inheritance and
phenotypes. Genes Brain Behav. 12:234–240. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Huang XJ, Wang T, Wang JL, Liu XL, Che XQ,
Li J, Mao X, Zhang M, Bi GH, Wu L, et al: Paroxysmal kinesigenic
dyskinesia: Clinical and genetic analyses of 110 patients.
Neurology. 85:1546–1553. 2015.PubMed/NCBI View Article : Google Scholar
|