Introduction
Ischemia-reperfusion (I-R) injury is implicated in
several human diseases, and consists of two phases; ischemia and
reperfusion. Occlusion of an artery or a decreased effective blood
volume results in the ischemic phase. During the ischemic period,
cell injury ensues as a result of anoxia-induced cellular energy
collapse (1-3).
Recanalization of the occluded artery or restoration of the
effective blood volume restores blood supply leading to the
reperfusion phase. During the reperfusion period, the re-entry of
oxygen to the ischemic tissue results in a burst of reactive oxygen
species (ROS) production and eventually in cell injury and death
(1-3).
Similar to other organs, such as the brain and the heart (1-3),
the kidney is extremely vulnerable to I-R injury, with this type of
injury being the leading cause of acute kidney injury (4). It has previously been shown that mouse
renal proximal tubular epithelial cells (RPTECs) die as a result of
apoptosis during anoxia, whereas during reoxygenation death ensues
from the increased ROS production, and the subsequent lipid
peroxidation-induced cell death, or otherwise ferroptosis (5).
The transcription factor nuclear factor erythroid
2-related factor 2 (Nrf2) regulates the transcription of several
antioxidant and anti-ferroptotic genes (6,7), and it
has been shown to protect against ferroptosis during reoxygenation.
This is particularly true in RPTECs derived from the hibernator
Syrian hamster (8,9). Hibernation involves cycles of torpor,
characterized by a notable decline in heart and breathing rates, as
well as in blood pressure, interrupted by interbout arousals and
restoration of the above parameters. Hence, unlike most mammals,
hibernators resist repeated cycles of I-R (10,11). In
our previous study, it was shown that Nrf2 is activated during
reoxygenation and is cytoprotective against ferroptosis in Syrian
hamster RPTECs subjected to warm anoxia and subsequent
reoxygenation (8). However, the
mechanism underlying an insufficiency of the above system to
protect non-hibernating mammals from reoxygenation-induced cell
death remains to be determined.
Hydrogen sulfide (H2S) serves a
significant role in Nrf2 activation. H2S is produced by
the enzymes cystathionine β-synthase (CBS), cystathionine γ-lyase
(CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST) (12). H2S interacts with, and
induces conformational changes in Kelch-like ECH-associated protein
1 (Keap1), resulting in the release of Nrf2 from the Nrf2-Keap1
complex and rescuing Nrf2 from proteasomal degradation (13,14).
Following release from Keap1, Nrf2 translocates to the nucleus and
increases transcription of several antioxidant genes (6,7). This
process is activated by reoxygenation in hibernating species
offering protection against cell injury. It has been shown that all
the aforementioned H2S-producing enzymes, as well as the
H2S levels, are upregulated during reoxygenation and
this results in the activation of Nrf2 in the context of resistance
to reoxygenation-induced cell death in Syrian hamster RPTECs
(8).
Mouse RPTECs die during anoxia through apoptosis
(5). The role of the H2S-Nfr2 axis
in apoptotic cell death is contradictory. Previous studies have
shown that endogenous H2S or Nrf2 activation protects
against apoptosis (15-17);
whereas other studies have shown that H2S induces
apoptosis (18-20).
One of the latter studies which showed H2S-induced
apoptosis, suggesting that apoptosis was mediated by the activation
of the proapoptotic p53/Bcl-2-associated X protein (Bax) pathway
(20).
In support of the protective role of the
H2S-Nrf2-antioxidant proteins axis, numerous studies
have shown that exogenous sulfide donors protect organs against I-R
injury (21-24),
including the kidneys (25-27).
To evaluate the possible role of the
H2S-Nrf2-antioxidant proteins axis in protecting
non-hibernator mammals against I-R injury, the kinetics of the
above axis in RPTECs derived from the non-hibernator mouse
subjected to anoxia or reoxygenation were assessed. The
non-specific inhibitor aminooxyacetate (AOAA) was used as a
H2S production inhibitor. AOAA directly inhibits CBS and
CSE (28), and 3-MST indirectly, as
3-MST converts cysteine to pyruvate with the assistance of cysteine
aminotransferase, and AOAA inhibits transamination (29). Additionally, when needed, the lipid
peroxidation and ferroptosis inhibitor α-tocopherol were used
(30).
Materials and methods
Cell culture and treatment
Primary C57BL/6 mouse RPTECs (cat. no. C57-6015,
Cell Biologics, Inc.) were cultured in Complete Epithelial Cell
Medium kit, supplemented with epithelial cell growth supplement
(0.1% epithelial growth factor, 0.1% insulin-transferrin-selenium
(ITS), 1% L-glutamine, 2% fetal bovine serum and 1% antibiotics)
(cat. no. M6621; Cell Biologics, Inc.). For all experiments, cells
were used after the second passage.
RPTECs were cultured in 96-well plates
(1x104 cells/well) or in 6-well plates (3x105
cells/well) at 37˚C. To simulate ischemia, cells were placed for 24
h in a GasPak™ EZ Anaerobe Container system with an on-board
methylene blue indicator tablet with a distinct color reaction. The
indicator remains colorless (white) under anaerobic conditions and
changes to blue once exposed to oxygen. (cat. no. 26001: BD
Biosciences). This system was used to ensure an oxygen
concentration <1%.
To simulate reperfusion, after 24 h of anoxia,
RPTECs were removed from the Anaerobe Container system and washed
with PBS (Sigma-Aldrich; Merck KGaA), the culture medium was
replaced with fresh rmedium, and the cells were cultured in a
humidified atmosphere containing 5% CO2 at 37˚C for 2
h.
The periods of anoxia and reoxygenation were
selected based on our previous study, in which it was shown that
primary mouse RPTECs viability declines considerably after 48 h of
anoxia and 4 h of reoxygenation (5).
Cells were harvested after 24 h of anoxia or after 2 h of
reoxygenation, as past these time points, the cell condition was
deteriorated considerably, with the majority of cells not being
suitable for further analysis (5).
Each experiment was repeated six times.
For evaluating ferroptosis, 100 µΜ of α-tocopherol
(Sigma-Aldrich; Merck KGaA) was added to inhibit lipid peroxidation
and ferroptosis. For assessing the effect of H2S, 2 mM
AOAA (Selleck Chemicals) was used to inhibit
H2S-producing enzymes. The above AOAA concentration was
selected after assessing its cytotoxicity in mouse RPTECs, as
described below.
AOAA cytotoxicity in RPTECs
Mouse RPTECs were cultured in 96-well plates in a
humidified atmosphere containing 5% CO2 in the
presence/absence of AOAA at a concentration of 0.5, 1 or 2 mM for
24 h. A lactate dehydrogenase (LDH) release assay was performed to
assess cytotoxicity using a Cytotox Non-Radioactive Cytotoxic assay
kit (Promega Corporation). Cell necrosis was calculated using the
following formula: Cell necrosis (%)=(LDH in the supernatant/total
LDH) x100. Experiments were repeated six times.
Evaluation of proteins of
interest
Mouse RPTECs were cultured in 6-well plates.
Following anoxia and/or reoxygenation, RPTECs were lysed using
T-PER tissue protein extraction reagent (Thermo Fisher Scientific
Inc.) supplemented with protease (Sigma-Aldrich; Merck KGaA) and
phosphatase inhibitors (Roche Diagnostics). After protein
quantification using a Bradford assay (Sigma-Aldrich; Merck KGaA),
10 µg of protein from each sample was used for western blotting.
Proteins were electrophoresed using a 4-12% bis-tris acrylamide
gels (cat. no. NP0323BOX, NuPAGE 4-12% Bis-Tris Gel 1 mm x 15 well;
Invitrogen; Thermo Fisher Scientific, Inc.). Proteins were
transferred to a PVDF membrane, and membranes were blocked using
skimmed milk in Tris-buffered saline with Tween-20. Blots were
incubated with the primary antibody against the protein of interest
for 16 h at 4˚C, followed by incubation with the secondary antibody
incubation for 30 min at room temperature. The LumiSensor Plus
Chemiluminescent HRP Substrate kit (GenScript) was used for
enhanced chemiluminescent detection of the bands. The Restore
Western Blot Stripping Buffer (Thermo Fisher Scientific Inc.) was
used whenever reprobing of the PVDF blots was required.
Densitometry analysis was performed using ImageJ version 1.51t
(National Institutes of Health). Experiments were repeated six
times.
Primary antibodies used were specific for CBS
(1:1,000; cat. no. TA338394; OriGene Technologies Inc.), CSE
(1:100; cat. no. sc-374249; Santa Cruz Biotechnology, Inc.), 3-MST
(1:100; cat. no. sc-376168; Santa Cruz Biotechnology, Inc.), Nrf2
(1:1,000; cat. no. TA343586; OriGene Technologies, Inc.),
superoxide dismutase 3 (SOD3; 1:100; cat. no. sc-271170; Santa Cruz
Biotechnology, Inc.), glutathione reductase (GR; 1:100; cat. no.
sc-133245; Santa Cruz Biotechnology, Inc.), ferritin heavy chain
(1:100; cat. no. sc-376594; Santa Cruz Biotechnology, Inc.),
cystine-glutamate antiporter (xCT; 1:1,000; cat. no. ANT-111;
Alomone Labs), activated cleaved-caspase-3(175) (1:500; cat. no.
7074; Cell Signaling Technology, Inc.), p53 (1:500; cat. no. 2524
Cell Signaling Technology, Inc.), p53 phosphorylated at serine 15
(p-p53) (1:500; cat. no. 9284; Cell Signaling Technology, Inc.),
Bax (1:500; cat. no. 5023; Cell Signaling Technology, Inc.) and
β-actin (1:2,500; cat. no. 4967; Cell Signaling Technology, Inc.).
Horseradish peroxidase-conjugated anti-rabbit IgG (1:1,000; cat.
no. 7074; Cell Signaling Technology, Inc.) or horseradish
peroxidase-conjugated anti-mouse IgG (1:1,000, cat. no. 7076; Cell
Signaling Technology, Inc.) were used as secondary antibodies.
Assessment of H2S
production
At the end of the 24-h anoxia period and the 2-h
reoxygenation periods, H2S production was assessed by
measuring its concentration in the supernatants of RPTECs cultured
in 6-well plates. The effect of the H2S-producing
enzymes inhibitor AOAA was also assessed. H2S production
was measured using a methylene blue assay as described previously
(31,32). Zinc acetate (1% w/v) (Sigma-Aldrich;
Merck KGaA) was added immediately to 1 ml of each supernatant to
trap the produced H2S. N,N-dimethyl-p-phenylenediamine
dihydrochloride (400 mg) (Sigma-Aldrich; Merck KGaA) was dissolved
in 10 ml 6 M HCl, ferric chloride (600 mg; Sigma-Aldrich; Merck
KGaA) in 10 ml 6 M HCl, and then, 1 ml from each of the two
solutions were mixed to prepare the required diamine-ferric
solution. Subsequently, 50 µl diamine-ferric solution was added to
each supernatant for 30 min and incubated at 37˚C, 200 µl of each
reaction was placed in the wells of a 96-well plate, and the amount
of methylene blue formed in each supernatant was measured at 670 nm
on an EnSpire® Multimode Plate Reader (PerkinElmer,
Inc.). To extrapolate the results of each experimental reaction,
different concentrations of methylene blue were also measured to
create a standard curve (Merck KGaA). These experiments were
repeated six times.
Evaluation of ROS production
ROS production was measured in RPTECs cultured in
96-well plates. Following anoxia and reoxygenation, 5 µM
CellROX® Deep Red Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), a fluorogenic probe, was added to the culture
medium and cells were incubated at 37˚C for 30 min. Subsequently,
RPTECs were washed with PBS, and an EnSpire® Multimode
Plate Reader was used to measure fluorescence signal intensity.
These experiments were repeated six times.
Assessment of lipid peroxidation
Lipid peroxidation was assessed in RPTECs cultured
in 6-well-plates. Malondialdehyde (MDA), the end-product of lipid
peroxidation, was measured fluorometrically in cell extracts with a
Lipid Peroxidation (MDA) assay kit (cat. no. ab118970; Abcam). The
kit fluorometrically detects MDA levels as low as 0.1 nmol. A
Bradford assay was performed prior to MDA measurement, and the
lysate volumes of all samples were adjusted to a protein
concentration of 1 mg/ml. These experiments were repeated six
times.
Assessment of cell ferroptosis
In mouse RPTECs subjected to reoxygenation, cell
death ensues via ferroptosis. To assess ferroptosis, at the end of
the reoxygenation period, cell necrosis was assessed in RPTECs
cultured in 96-well plates, in the presence/absence of lipid
peroxidation and ferroptosis inhibitor α-tocopherol. The effects of
the H2S-producing enzymes inhibitor AOAA was also
evaluated. An LDH release assay was performed for assessing cell
necrosis using the Cytotox Non-Radioactive Cytotoxic assay kit.
Cell necrosis was calculated as follows: Cell necrosis (%)=(LDH in
the supernatant/total LDH)x100. These experiments were repeated six
times.
Statistical analysis
SPSS Version 20 (IBM, Corp.) was used for
statistical analysis. A one-sample Kolmogorov-Smirnov test was used
to confirm that the evaluated variables were normally distributed.
For comparison of means, a one-way ANOVA followed by Bonferroni's
correction test was used. Results are expressed as the mean ±
standard error of mean. P<0.05 was considered to indicate a
statistically significant difference.
Results
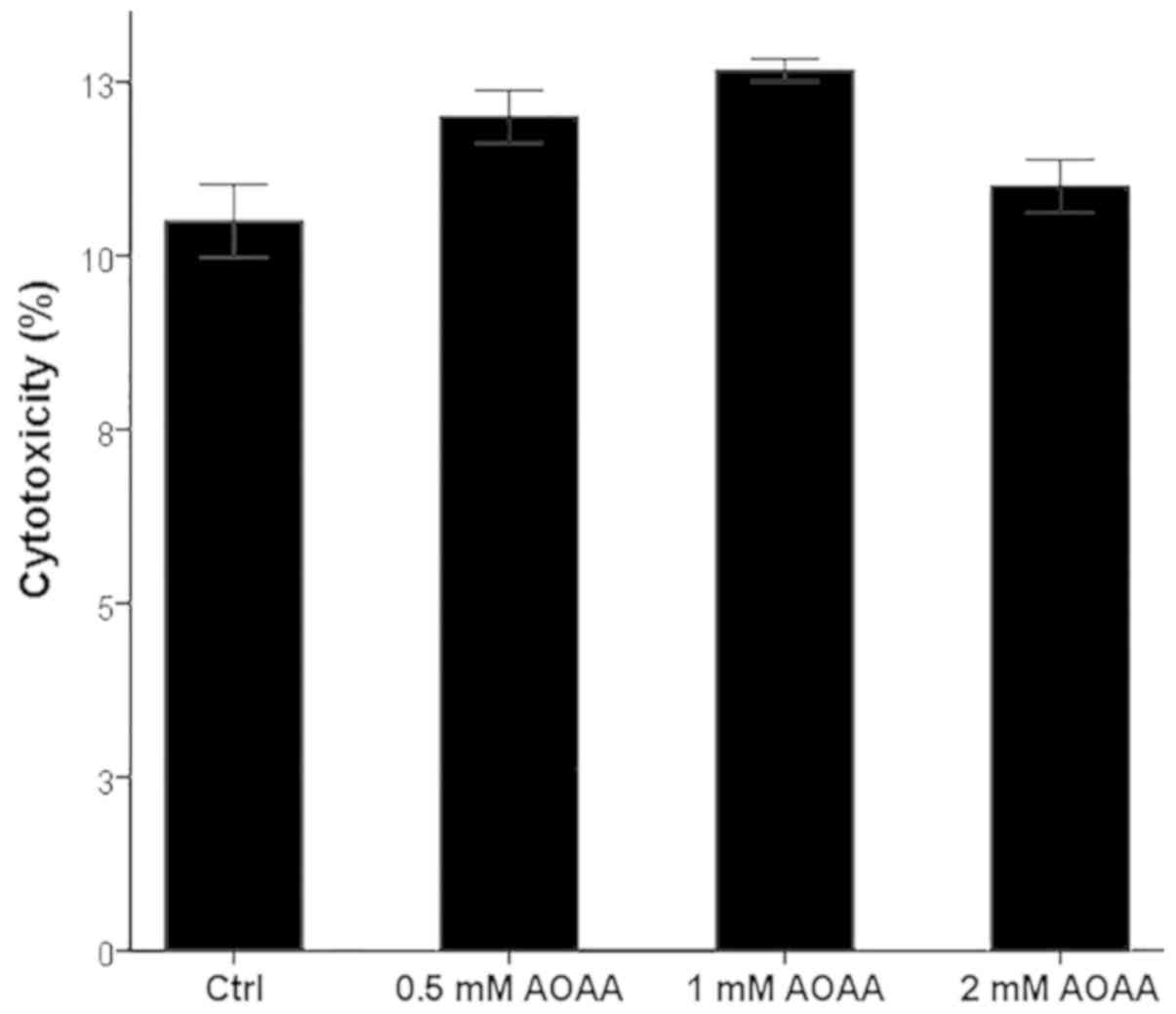
AOAA is not toxic for RPTECs at the
assessed concentrations
AOAA did not exhibit any notable cytotoxic effects
at any of the tested concentrations (Fig. 1). A concentration of 2 mM was
selected for all subsequent experiments.
ROS production, lipid peroxidation,
ferroptosis and the role of endogenous H2S
Compared with the control RPTECs, in RPTECs cultured
under anoxic conditions, ROS production did not differ
significantly (signal intensity 40.4±0.4 vs. 39.6±1.5,
respectively). However, reoxygenation increased ROS production
significantly (signal intensity 71.0±3.2; P<0.001 compared with
control and anoxia; Fig. 2A).
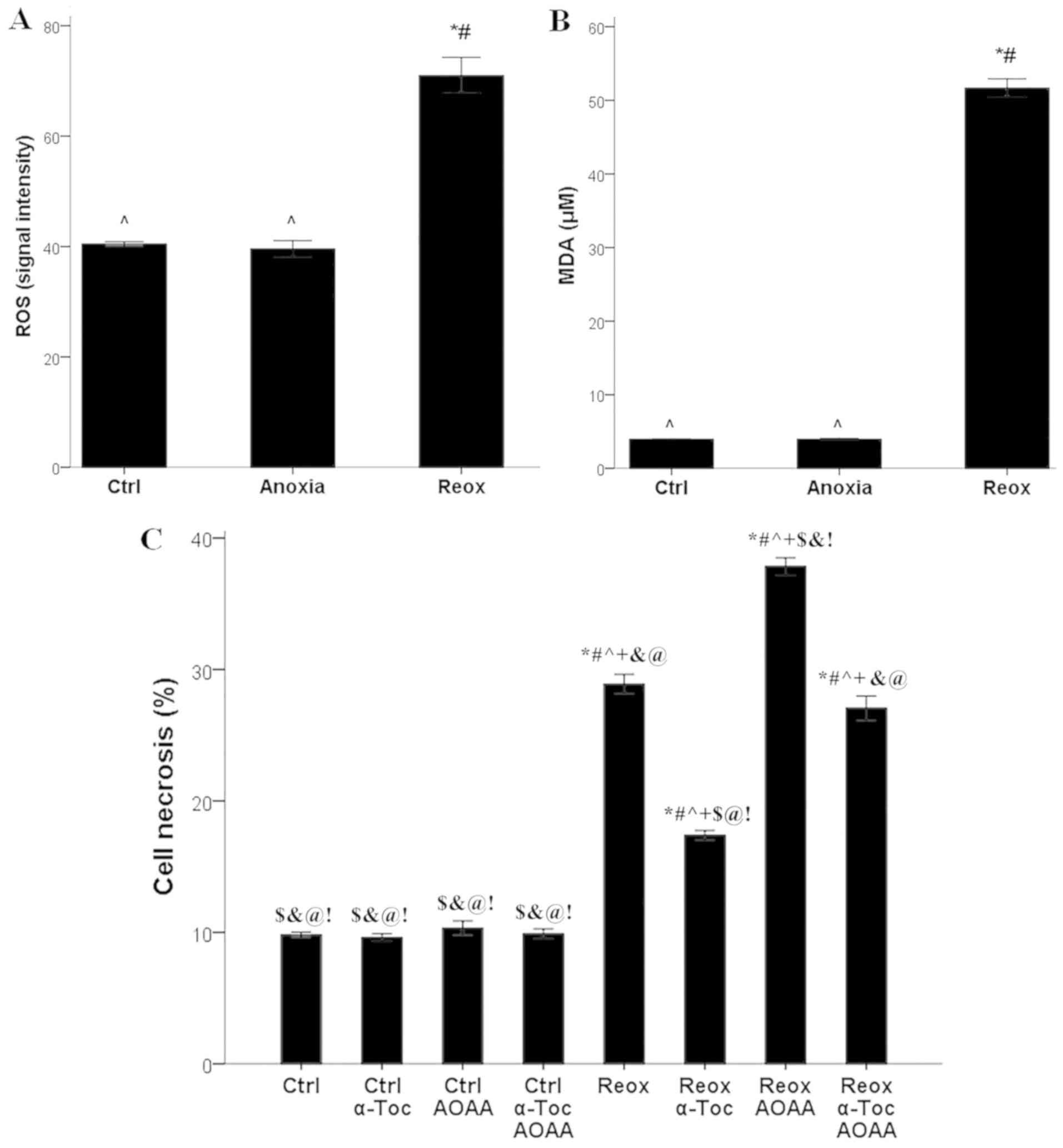 | Figure 2ROS production, lipid peroxidation,
cell necrosis and the effect of AOAA and α-tocopherol. (A) Only
reoxygenation increased ROS production, (B) lipid peroxidation,
based on the levels of MDA. *P<0.001 vs. Ctrl;
#P<0.001 vs. Anoxia; ^P<0.001 vs. Reox.
(C) Reoxygenation induced cell necrosis. α-tocopherol protected
renal proximal tubular epithelial cells from reoxygenation-induced
cell necrosis, whereas AOAA aggravated cell necrosis.
*P<0.001 vs. Ctrl; #P<0.001 vs. Ctrl +
α-Toc; ^P<0.001 vs. Ctrl + AOAA;
+P<0.001 vs. Ctrl + α-Toc + AOAA;
$P<0.001 vs. Reox; &P<0.001 vs.
Reox + α-Toc; @P<0.001 vs. Reox + AOAA;
!P<0.001 vs. Reox + α-Toc + AOAA. ROS, reactive
oxygen species; MDA, malondialdehyde; AOAA, aminooxyacetate; Ctrl,
control; α-Toc, α-tocopherol; Reox, reoxygenation. |
Anoxia did not significantly affect lipid
peroxidation, whereas reoxygenation induced lipid peroxidation in
RPTECs. MDA levels were 4.0±0.1 µM in control cells and 4.0±0.1 µM
in RPTECs cultured under anoxic conditions. Reoxygenation increased
MDA levels significantly to 51.7±1.2 µM (P<0.001 compared with
control and anoxia; Fig. 2B).
In RPTECs cultured under normoxic conditions, the
ferroptosis inhibitor α-tocopherol, the H2S-producing
enzymes inhibitor AOAA, or their combination did not significantly
alter cell necrosis, which was 9.8±0.2, 9.6±0.3, 10.3±0.6 and
9.9±0.4%, respectively. Reoxygenation increased cell necrosis to
28.8±0.7% (P<0.001 compared with control; Fig. 2C).
Treatment of RPTECs subjected to reoxygenation with
α-tocopherol ameliorated cell necrosis considerably (17.4±0.4%;
P<0.001 compared with reoxygenation alone), whereas treatment
with AOAA significantly increased cell necrosis (37.8±0.9%;
P<0.001 compared with reoxygenation alone; Fig. 2C).
Collectively, these results show that reoxygenation
increases ROS production and induces ferroptosis, and also suggests
a protective role of endogenous H2S against
ferroptosis.
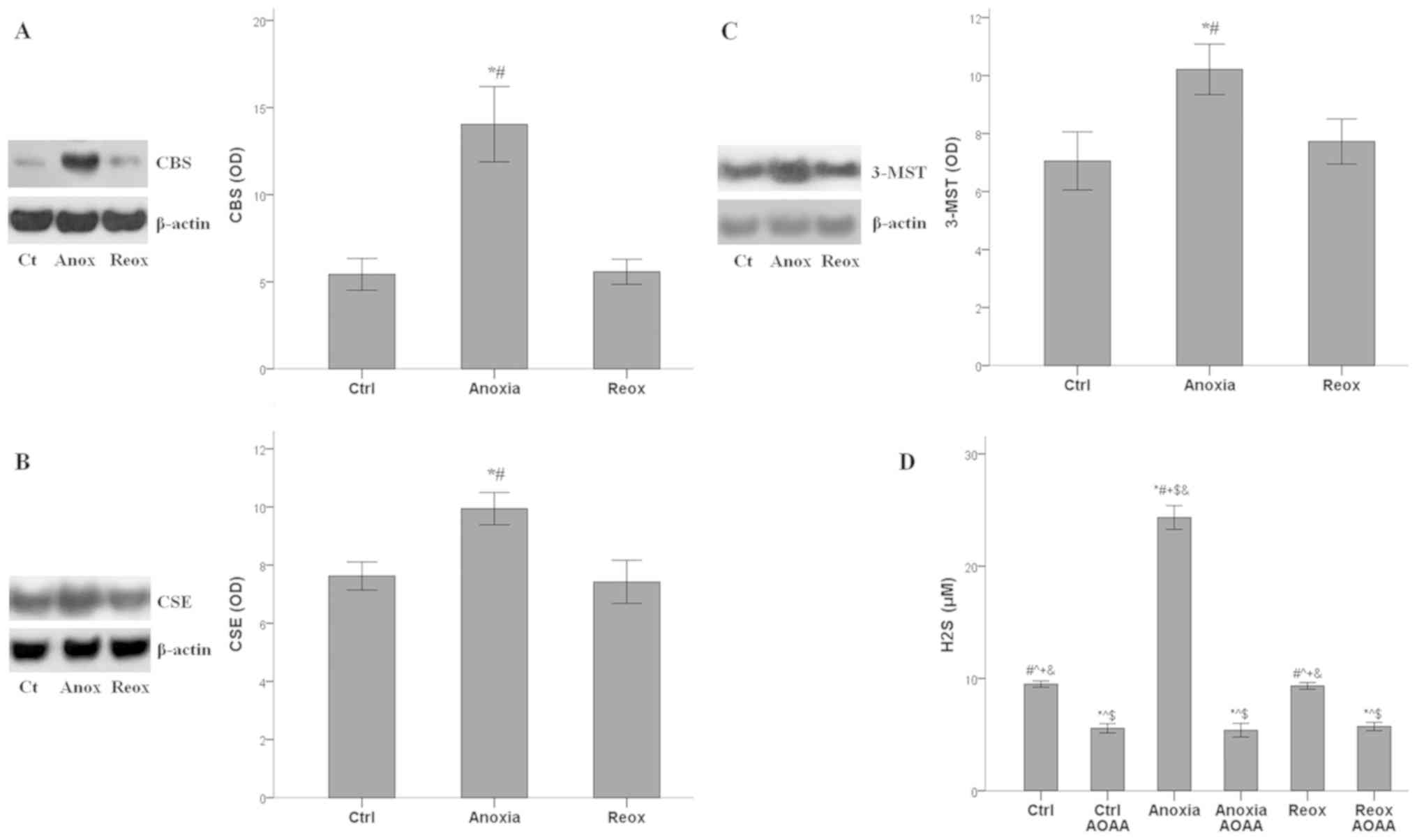
Kinetics of H2S-producing enzymes
expression and H2S under anoxia and reoxygenation
The expression of all the H2S-producing
enzymes increased significantly under anoxia (P<0.001 compared
with the control) and returned to baseline during reoxygenation.
For CBS, the mean optical density (OD) was 5.4±0.9 under normoxia,
14.0±2.2 under anoxia and 5.6±0.7 under reoxygenation (Fig. 3A). The OD values of CSE were 7.6±0.5,
10.0±0.6 and 7.4±0.7 under normoxia, anoxia and reoxygenation,
respectively (Fig. 3B). For 3-MST,
the ODs were 7.1±1.0, 10.2±0.9 and 7.7±0.8, under normoxia, anoxia
and reoxygenation, respectively (Fig.
3C).
Production of H2S follows the pattern of
H2S-producing enzymes expression. Under control
conditions, H2S concentration was 9.5±0.3 µM, under
anoxia it increased to 24.3±1.1 µM (P<0.001 compared with the
control), and reoxygenation decreased H2S concentration
to 9.3±0.3 µM (P>0.05 compared with the control). The presence
of AOAA reduced the H2S concentration significantly
under all conditions. In control cells, AOAA decreased
H2S concentration to 5.6±0.4 µM (P<0.001 compared
with the control alone), in RPTECs under anoxia to 5.4±0.6 mM
(P<0.001 compared with anoxia alone), and in RPTECs subjected to
reoxygenation to 5.7±0.4 µM (P<0.001 compared with reoxygenation
alone; Fig. 3D).
Hence, in RPTECs, all H2S-producing
enzymes, and thus H2S production, were upregulated
during anoxia and returned to baseline during reoxygenation. AOAA
decreased H2S production under all cell culture
conditions.
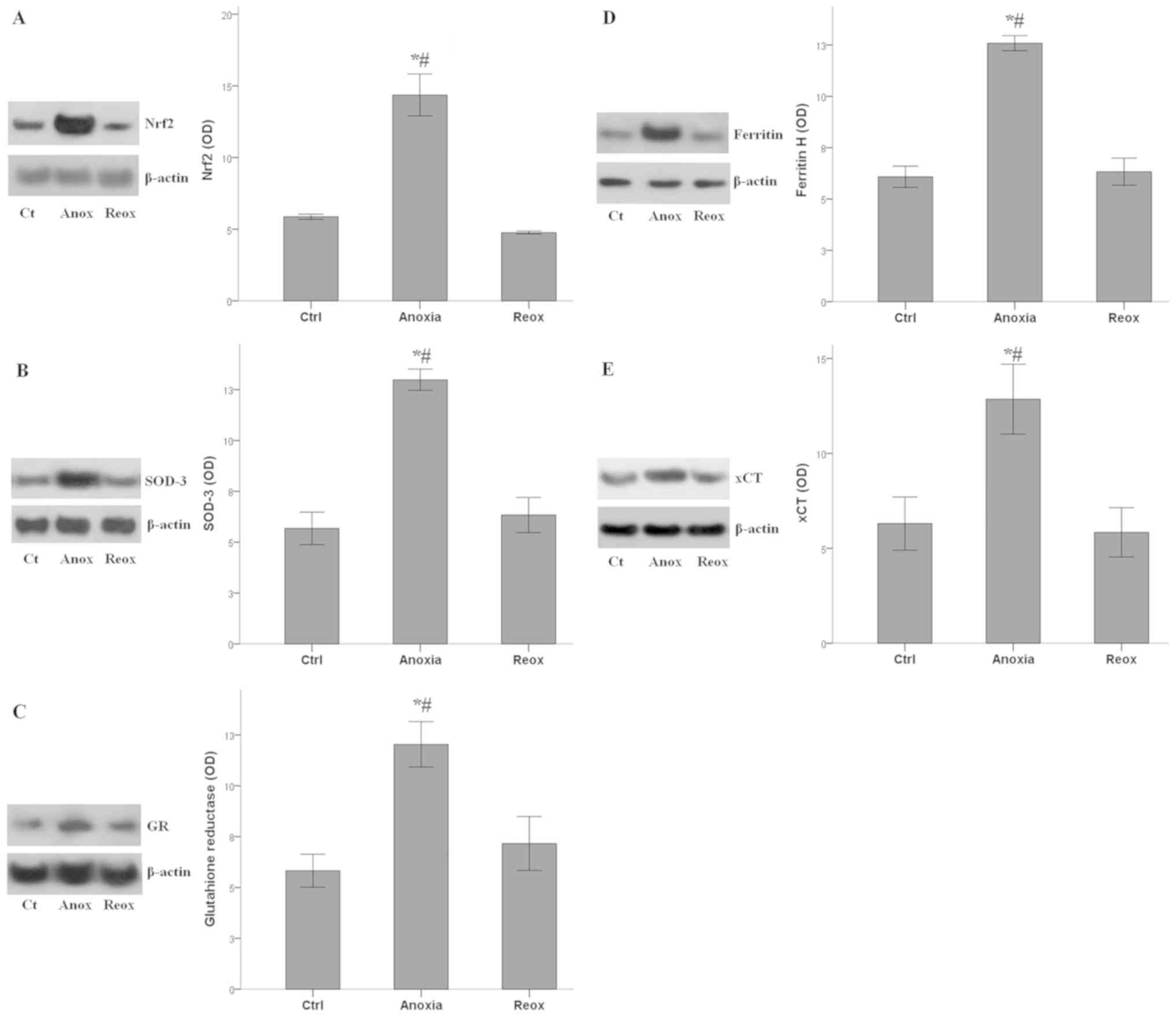
Activation status of Nrf2-antioxidant
proteins axis during anoxia and reoxygenation
The Nrf2 levels followed the fluctuations of
H2S concentrations under anoxia and reoxygenation. Nrf2
mean OD was 5.9±0.2 under control conditions, which increased to
14.4±1.5 under anoxia (P<0.001 compared with the control), and
returned to baseline under reoxygenation (4.8±0.1; P>0.05
compared with the control; Fig.
4A).
The expression of all the evaluated antioxidant
proteins, which are transcriptional targets of Nrf2, followed the
fluctuations of Nrf2 levels. SOD-3 OD was 5.7±0.8 under control
conditions, which increased to 13.0±0.5 under anoxia (P<0.001
compared with the control), and returned to baseline under
reoxygenation (6.3±0.9; P>0.05 compared with the control;
Fig. 4B). OD values for GR were
5.8±0.8 under normal conditions, 12.0±1.1 under anoxia (P<0.001
compared with the control) and 7.2±1.3 under reoxygenation
(P>0.05 compared with the control; Fig. 4C). For Ferritin H the OD values were
6.1±0.5 under normal conditions, 12.6±0.4 under anoxia (P<0.001
compared with the control) and 6.3±0.7 under reoxygenation
(P>0.05 compared with the control; Fig. 4D). Finally, for xCT the OD values
were 6.3±1.4 under normal conditions, 12.9±1.8 under anoxia
(P<0.001 compared with the control) and 5.8±1.3 under
reoxygenation (P>0.05 compared with the control; Fig. 4E).
Therefore, in RPTECs, the anoxia-induced increase in
H2S production enhanced both Nrf2 levels and the
expression of various antioxidant proteins, which is under the
transcriptional control of Nrf2. During reoxygenation, when ROS
levels increase and when the antioxidant proteins are required, the
axis remained inactive.
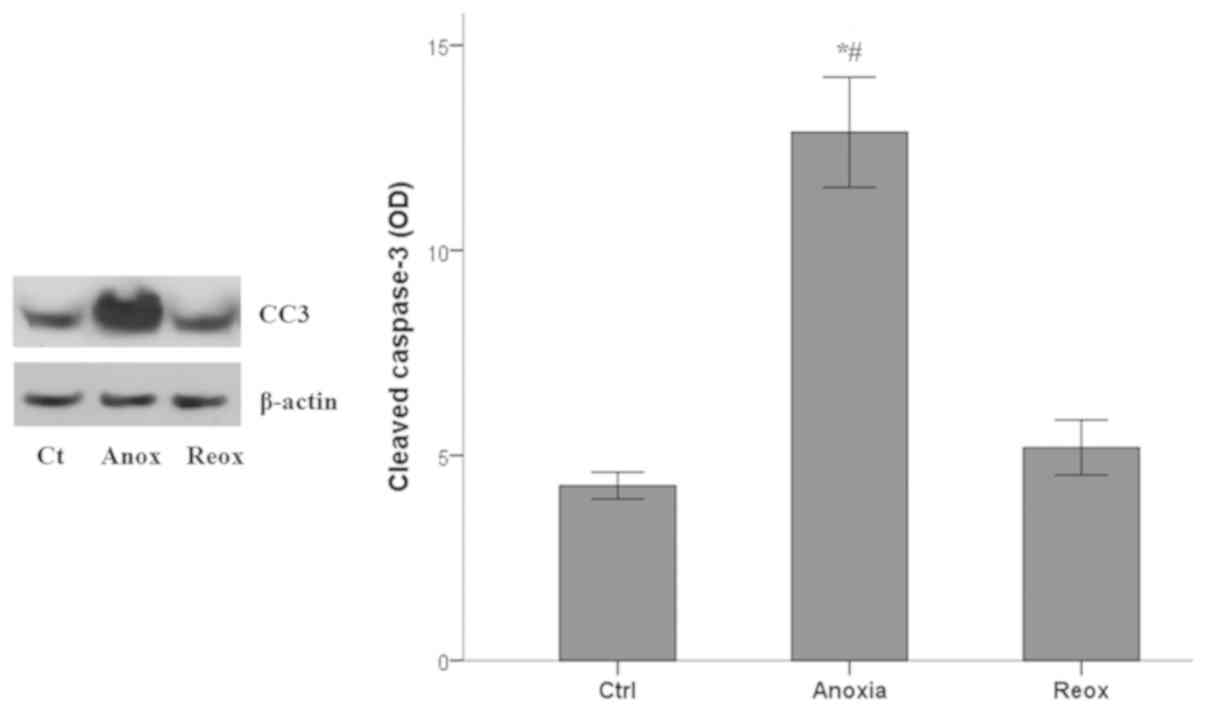
Apoptosis during anoxia and
reoxygenation
Apoptotic cell death was assessed by measuring the
levels of activated cleaved-caspase-3, in which all the apoptotic
pathways converge (33). Anoxia
induced apoptosis in RPTECs, whereas reoxygenation did not.
Cleaved-caspase-3 mean OD values 4.3±0.3 under control conditions,
increased to 12.9±1.3 under anoxia (P<0.001 compared with the
control) and returned to baseline levels (5.2±0.7; P>0.05
compared with the control) in RPTECs subjected to reoxygenation
(Fig. 5).
Thus, in mouse RPTECs, apoptosis ensues only under
anoxic conditions and not during the reoxygenation phase.
Effect of H2S on
anoxia-induced apoptosis and on the pro-apoptotic p53-Bax axis
Since anoxia induces apoptosis in RPTECs, and
various studies have produced contradictory results regarding the
effects of endogenous H2S on apoptosis, and on the
proapoptotic p53-Bax axis, the impact of the
H2S-producing enzymes inhibitor AOAA on the above
parameters in RPTECs subjected to anoxia were assessed.
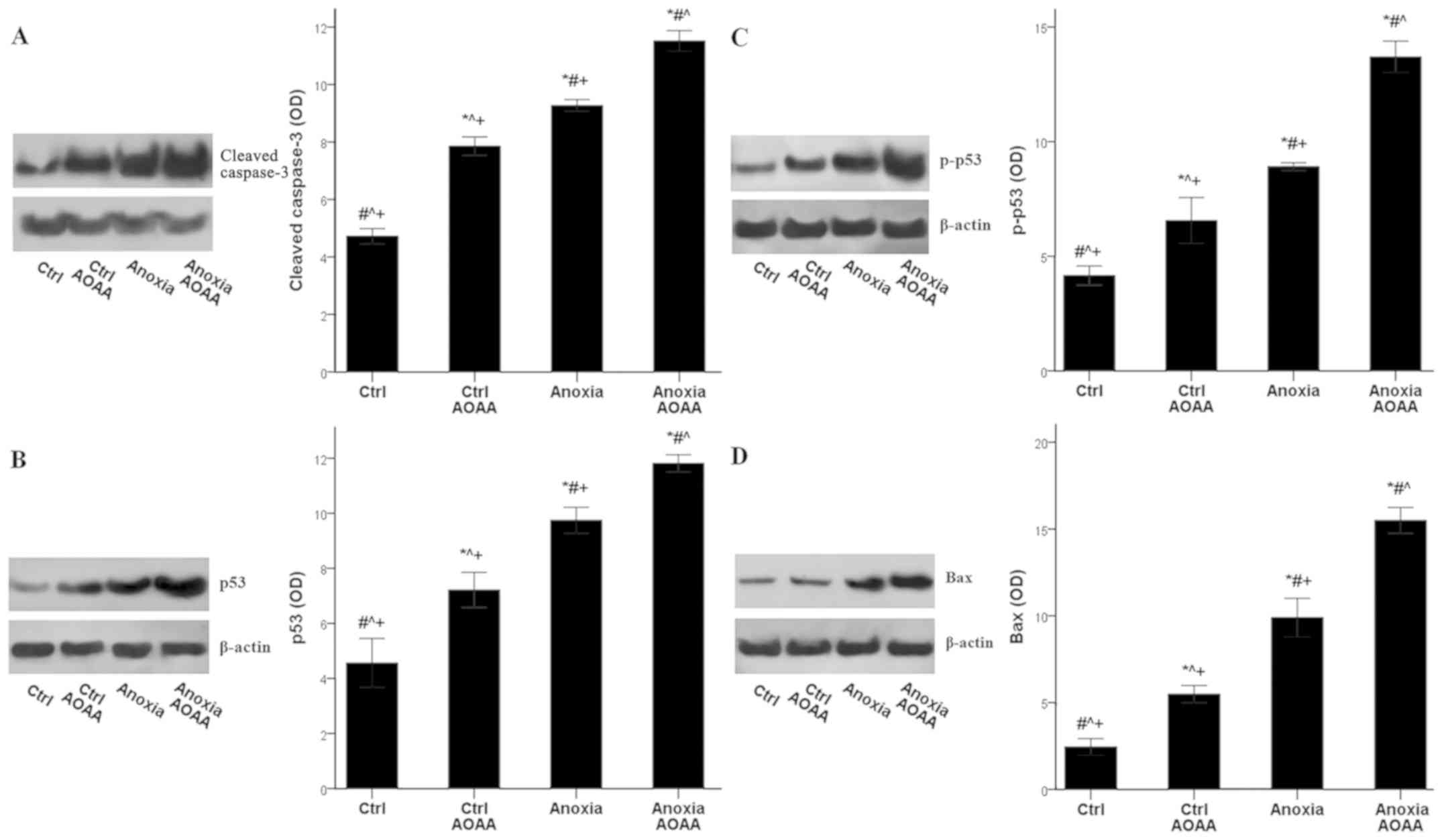
In RPTECs subjected to anoxia, AOAA increased
apoptosis further. The mean OD values of the cleaved-caspase-3 was
4.7±0.3 in the control RPTECs, which increased to 9.3±0.2 in RPTECs
subjected to anoxia (P<0.001 compared with the control), and
increased even further to 11.5±0.4 in RPTECs subjected to anoxia
and treated with AOAA (P<0.001 compared with the control and
anoxia). Additionally, under control conditions, AOAA enhanced
cleaved-caspase-3 levels to 7.9±0.3 (P<0.001 compared with the
control; Fig. 6A).
The increase in apoptosis due to AOAA treatment may
result from the activation of the proapoptotic p53-Bax axis. The
p53 OD values were 4.6±0.9 in control RPTECs, increased to 9.7±0.5
under anoxia (P<0.001 compared with the control), and further
increased to 11.8±0.3 in the presence of AOAA (P<0.001 compared
with the control and anoxia). Additionally, AOAA enhanced p53
levels to 9.7±0.3 under control conditions (P<0.001 compared
with the control; Fig. 6B).
These alterations in p53 levels were likely the
result of an increase in its phosphorylation, which dissociates p53
from mouse double minute 2 homolog (MDM2) and protects p53 from
proteasomal degradation. The p-p53 OD was 4.2±0.4 in control
RPTECs, which increased to 8.9±0.2 under anoxia (P<0.001
compared with the control), and further to 13.7±0.7 when treated
with AOAA (P<0.001 compared with the control and anoxia). AOAA
treatment increased the p-p53 OD to 6.6±1.0 in RPTECs cultured
under control conditions (P<0.001 compared with the control;
Fig. 6C).
The expression of the proapoptotic protein Bax
followed the pattern of p53, which controls Bax transcription.
Anoxia increased Bax OD values from 2.4±0.5 in the control to
9.9±1.1 under anoxia (P<0.001), and anoxia combined with AOAA
treatment further increased Bax levels (15.5±0.7; P<0.001
compared with the control and anoxia). Treatment of RPTECs with
AOAA under normoxia also increased Bax expression levels (5.5±0.5;
P<0.001 compared with the control; Fig. 6D).
Based on these results, it is hypothesized that
endogenous H2S, which increases under anoxic conditions,
ameliorates anoxia-induced apoptosis by interfering with the
p53-Bax axis. However, as the anoxia-induced changes proceed,
apoptosis occurs indicating that the upregulation in
H2S-producing enzymes during anoxia is insufficient for
preventing apoptosis completely.
Discussion
In mouse RPTECs, ROS production was increased only
during reoxygenation, when oxygen was available, resulting in lipid
peroxidation and cell necrosis. The inhibitor of lipid peroxidation
and ferroptosis, α-tocopherol, protected mouse RPTECs against
reoxygenation-induced cell necrosis, indicating that during this
phase, cell death occurs via ferroptosis (34). Similar results were obtained in a
previous study with isolated mouse renal tubules subjected to
anoxia and subsequent reoxygenation (35). Treatment with AOAA increased
ferroptosis, underlying the protective role of H2S.
Expression of all the assessed H2S
producing enzymes, as well as H2S levels, were
upregulated during anoxia; the increased H2S levels
upregulated Nrf2 expression levels, and thus indirectly, the
expression of the assessed transcriptional targets of Nrf2
(6,7,13,14). The
targets assessed were SOD-3, which catalyzes the dismutation of
superoxide radicals into either molecular oxygen or hydrogen
peroxide (36); GR, which catalyzes
the reduction of glutathione disulfide to the sulfhydryl form
glutathione (GSH) (36); ferritin,
which functions by sequestering intracellular labile iron
preventing the Fenton reaction and blocking the function of
iron-containing lipoxygenases (30);
and the cystine-glutamate antiporter xCT, which is responsible for
the entry of the cystine required for GSH synthesis into the cell
and prevents ferroptosis (30). All
the above Nrf-2 targets were upregulated during anoxia.
However, during reoxygenation, when the activation
of the antioxidant defense system is required due to ROS
overproduction, the H2S producing
enzymes-Nrf2-antioxidant proteins axis was downregulated to
baseline levels, leaving the cells unprotected. Thus, during
anoxia-reoxygenation injury, this mistimed activation of the above
axis is unable to protect RPTECs against reoxygenation-induced
ferroptotic cell death. Previous studies confirmed that hypoxia
upregulates CBS, CSE and 3-MST levels (37-39).
However, the mechanisms involved in their downregulation during
reoxygenation remains to be elucidated; an understanding of the
underlying mechanisms may assist in the development of novel
therapeutic approaches for treating I-R injury.
The significance of the above axis in protecting
cells against reoxygenation-induced ferroptosis has been detected
in hibernating species, which are subjected to repeated cycles of
I-R during hibernation without experiencing any injuries (10,11).
Contrary to mouse RPTECs, in Syrian hamster RPTECs, the activation
of H2S producing enzymes-Nrf2-antioxidant proteins axis
takes place later, at the appropriate time during reoxygenation and
ROS overproduction, protecting cells from ferroptotic cell death
(8).
Furthermore, the results of the present study
support the possible protective role of exogenous sulfur donors in
the prevention of reoxygenation-induced cell death. Several studies
in non-hibernating species have demonstrated the protective effects
of such an intervention in experimental models of acute kidney
injury (25-27),
myocardial infarction (21),
cerebral stroke (22) and multiorgan
failure (23,24).
Since oxidative stress is implicated in several
renal pathologies, Nrf2 activators other than H2S donors
have also been assessed in experimental models of various kidney
diseases with promising results, and several clinical studies are
being performed (40). In an
experimental model of I-R-induced kidney injury, the Nrf2 activator
bardoxolone methyl ameliorated structural injury and renal
dysfunction in mice (41).
Bardoxolone imidazolide was also found to improve survival, renal
function, kidney histology and production of pro-inflammatory
cytokines in mice subjected to kidney I-R injury (42). Similar beneficial results were
observed in mice administered omaveloxolone, another Nrf2 activator
(43). Also, the interest in various
natural products able to activate Nrf2 is growing. For example, the
Nrf2 activator curcumin, was beneficial in a rat model of kidney
I-R injury (44), and total
flavonoids from Rosa laevigata Michx fruit upregulated Nrf2
expression, and exhibited beneficial effects in mice subjected in
kidney I-R injury (45).
The results of the present study suggested that
apoptosis, based on the levels of cleaved-caspase-3 in which all
the apoptotic pathways converge (33), was only observed during anoxia, and
not during reoxygenation. This is in agreement with the results of
a previous study in which mouse RPTECs were subjected to similar
experimental conditions (5).
Interestingly, in RPTECs under anoxia, administration of AOAA
decreased H2S production, and in parallel, aggravated
apoptotic cell death.
Under anoxia, in RPTECs the levels of
cleaved-caspase-3, p53 and Bax also increased, suggesting that
apoptosis may be mediated by the p53-Bax pro-apoptotic pathway
(46). The role of the p53-Bax
pathway in I-R injury has been established, and in a previous
study, transient silencing of p53 with a specific siRNA, protected
rat kidney function from I-R injury by preventing apoptosis of
RPTECs (47). The results of two
ongoing related clinical trials using a p53-specific siRNA; a phase
2 clinical trial on preventing acute kidney injury following
cardiac surgery (clinicaltrials.gov/ct2/show/results/NCT02610283), and
a phase 3 clinical trial on preventing delayed graft function
following kidney transplantation from old donors (clinicaltrials.gov/ct2/show/results/NCT02610296) will
clarify the clinical significance of p53 in I-R-induced
apoptosis.
The levels of p-p53 were also increased during
anoxia, suggesting that the increase in p53 levels was the result
of p53 dissociation from the ubiquitin ligase MDM2(46). The enhanced phosphorylation of p53
may result from anoxia-induced DNA damage, and the activation of a
group of kinases which are implicated in the genome integrity
checkpoint (46).
AOAA treatment resulted in an increase in the levels
of p-p53, p53, Bax and CC3, suggesting a protective role of the
H2S-Nrf2 axis against apoptosis. Interestingly, there is
a bidirectional interaction between Nrf2 and the p53 pathway. Nrf2
transcribes MDM2, which acts as a specific ubiquitin ligase and
downregulates p53. p53 directly suppresses Nrf2, although p53 also
transcribes p21, which is known to upregulate Nrf2(48). The exact molecular interactions that
transpire during anoxia remains to be elucidated.
The results of the present study are in agreement
with studies showing an anti-apoptotic role of the
H2S-Nrf2 axis (15-17),
contradicting studies which showed the opposite outcomes (18-20).
However, by interpreting the outcome of the experiments, which
indicate that despite anoxia-induced activation of the
H2S-Nrf2 axis, apoptosis occurs, it is likely that this
system by itself is unable to confer full protection on RPTECs
against anoxia-induced apoptosis. The latter highlights the
possibility of exogenous sulfur donors to protect RPTECs against
anoxia-induced apoptotic cell death. Combinations of sulfur donors
with apoptosis inhibitors may prove more effective in the
prevention or amelioration of I-R injury. Interestingly, and
contrary to the mouse RPTECs, in RPTECs derived from the hibernator
Syrian hamster, which resists apoptosis, the H2S-Nrf2
axis is activated at a later stage, during reoxygenation,
suggesting that in hibernating species, the above axis is not
responsible for the evident resistance to anoxia-induced apoptosis
(8,9).
A limitation of the present study is the in
vitro nature of the experiments. However, the strictly
controlled experimental conditions allowed the study of the two
different, subsequent, but distinct components of I-R injury
separately, and to assess the different kinetics of the
H2S producing enzymes-Nrf2-antioxidant proteins axis
under anoxia and reoxygenation, as well as its effect on cell
survival. Thus, our results may be considered a starting point for
further studies on the molecular mechanisms that govern the
activity of the above axis under anoxia and reoxygenation, as well
as for interventional in vivo studies.
In conclusion, the results of the present study
suggest that in RPTECs, the H2S-Nrf2 axis is activated
by anoxia, and although it ameliorates apoptosis, it does not
completely prevent apoptotic cell death, and is eventually
overwhelmed. On the contrary, under reoxygenation, when the sudden
increase in ROS production occurs, the antioxidant defense is
essential for the protection of cells against ferroptotic cell
death, the H2S producing enzymes-Nrf2-antioxidant
proteins axis is not upregulated. This mistimed activation of the
above axis contributes to reoxygenation-induced cell death.
Clarifying the precise molecular mechanisms underlying the mistimed
H2S producing enzymes-Nrf2-antioxidant proteins axis
activation may result in clinically useful interventions for
preventing I-R injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contribution
TE designed the study. GP and TE performed the
experiments. TE, GP, EN, GF, VL and IS analyzed the results. TE and
GP wrote the manuscript. All authors approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017(7018393)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bakthavachalam P and Shanmugam PST:
Mitochondrial dysfunction - Silent killer in cerebral ischemia. J
Neurol Sci. 375:417–423. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tsukamoto T, Chanthaphavong RS and Pape
HC: Current theories on the pathophysiology of multiple organ
failure after trauma. Injury. 41:21–26. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Cell death patterns due to warm
ischemia or reperfusion in renal tubular epithelial cells
originating from human, mouse, or the native hibernator hamster.
Biology (Basel). 7(E48)2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ma Q: Role of Nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Corsello T, Komaravelli N and Casola A:
Role of hydrogen sulfide in NRF2- and sirtuin-dependent maintenance
of cellular redox balance. Antioxidants (Basel).
7(E129)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Eleftheriadis T, Pissas G, Nikolaou E,
Liakopoulos V and Stefanidis I: The H2S-Nrf2-antioxidant proteins
axis protects renal tubular epithelial cells of the native
hibernator syrian hamster from reoxygenation-induced cell death.
Biology (Basel). 8(74)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Eleftheriadis T, Pissas G, Liakopoulos V
and Stefanidis I: Factors that may protect the native hibernator
syrian hamster renal tubular epithelial cells from ferroptosis due
to warm anoxia-reoxygenation. Biology (Basel).
8(E22)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Carey HV, Andrews MT and Martin SL:
Mammalian hibernation: Cellular and molecular responses to
depressed metabolism and low temperature. Physiol Rev.
83:1153–1181. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Storey KB and Storey JM: Metabolic rate
depression: The biochemistry of mammalian hibernation. Adv Clin
Chem. 52:77–108. 2010.PubMed/NCBI
|
|
12
|
Powell CR, Dillon KM and Matson JB: A
review of hydrogen sulfide (H2S) donors: Chemistry and potential
therapeutic applications. Biochem Pharmacol. 149:110–123.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang G, Zhao K, Ju Y, Mani S, Cao Q,
Puukila S, Khaper N, Wu L and Wang R: Hydrogen sulfide protects
against cellular senescence via S-sulfhydration of keap1 and
activation of Nrf2. Antioxid Redox Signal. 18:1906–1919.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hourihan JM, Kenna JG and Hayes JD: The
gasotransmitter hydrogen sulfide induces Nrf2-target genes by
inactivating the keap1 ubiquitin ligase substrate adaptor through
formation of a disulfide bond between Cys-226 and Cys-613. Antioxid
Redox Signal. 19:465–481. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Niture SK and Jaiswal AK: Nrf2 protein
up-regulates antiapoptotic protein Bcl-2 and prevents cellular
apoptosis. J Biol Chem. 287:9873–9886. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Niture SK and Jaiswal AK: Nrf2-induced
antiapoptotic Bcl-xL protein enhances cell survival and drug
resistance. Free Radic Biol Med. 57:119–131. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang TX, Shi XY and Liu YH: Endogenous
cystathionine-gamma-lyase/hydrogen sulfide pathway regulates
apoptosis of HepG2 cells. Yao Xue Xue Bao. 48:1233–1240.
2013.PubMed/NCBI
|
|
18
|
Yang G, Sun X and Wang R: Hydrogen
sulfide-induced apoptosis of human aorta smooth muscle cells via
the activation of mitogen-activated protein kinases and caspase-3.
FASEB J. 18:1782–1784. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Breza J Jr, Soltysova A, Hudecova S,
Penesova A, Szadvari I, Babula P, Chovancova B, Lencesova L, Pos O,
Breza J, et al: Endogenous H2S producing enzymes are involved in
apoptosis induction in clear cell renal cell carcinoma. BMC Cancer.
18(591)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ma K, Liu Y, Zhu Q, Liu CH, Duan JL, Tan
BK and Zhu YZ: H2S Donor, S-propargyl-cysteine, increases CSE in
SGC-7901 and cancer-induced mice: Evidence for a novel anti-cancer
effect of endogenous H2S? Plos One. 6(e20525)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ji K, Xue L, Cheng J and Bai Y:
Preconditioning of H 2 S inhalation protects against cerebral
ischemia/reperfusion injury by induction of HSP70 through
PI3K/Akt/Nrf2 pathway. Brain Res Bull. 121:68–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ganster F, Burban M, de la Bourdonnaye M,
Fizanne L, Douay O, Loufrani L, Mercat A, Calès P, Radermacher P,
Henrion D, et al: Effects of hydrogen sulfide on hemodynamics,
inflammatory response and oxidative stress during resuscitated
hemorrhagic shock in rats. Crit Care. 14(R165)2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Satterly SA, Salgar S, Hoffer Z, Hempel J,
DeHart MJ, Wingerd M, Raywin H, Stallings JD and Martin M: Hydrogen
sulfide improves resuscitation via non-hibernatory mechanisms in a
porcine shock model. J Surg Res. 199:197–210. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Han SJ, Kim JI, Park JW and Park KM:
Hydrogen sulfide accelerates the recovery of kidney tubules after
renal ischemia/reperfusion injury. Nephrol Dial Transplant.
30:1497–1506. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sekijima M, Sahara H, Miki K, Villani V,
Ariyoshi Y, Iwanaga T, Tomita Y and Yamada K: Hydrogen sulfide
prevents renal ischemia-reperfusion injury in CLAWN miniature
swine. J Surg Res. 219:165–172. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Xu Z, Prathapasinghe G, Wu N, Hwang SY,
Siow YL and O K: Ischemia-reperfusion reduces
cystathionine-beta-synthase-mediated hydrogen sulfide generation in
the kidney. Am J Physiol Renal Physiol. 297:F27–F35.
2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Asimakopoulou A, Panopoulos P, Chasapis
CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA
and Papapetropoulos A: Selectivity of commonly used pharmacological
inhibitors for cystathionine β synthase (CBS) and cystathionine γ
lyase (CSE). Br J Pharmacol. 169:922–932. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Miyamoto R, Otsuguro K, Yamaguchi S and
Ito S: Contribution of cysteine aminotransferase and
mercaptopyruvate sulfurtransferase to hydrogen sulfide production
in peripheral neurons. J Neurochem. 130:29–40. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Stipanuk MH and Beck PW: Characterization
of the enzymic capacity for cysteine desulphhydration in liver and
kidney of the rat. Biochem J. 206:267–277. 1982.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Talaei F, Bouma HR, Van der Graaf AC,
Strijkstra AM, Schmidt M and Henning RH: Serotonin and dopamine
protect from hypothermia/rewarming damage through the CBS/H2S
pathway. PLoS One. 6(e22568)2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176.
2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841.
2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fang YZ, Yang S and Wu G: Free radicals,
antioxidants, and nutrition. Nutrition. 18:872–879. 2002.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Takano N, Peng YJ, Kumar GK, Luo W, Hu H,
Shimoda LA, Suematsu M, Prabhakar NR and Semenza GL:
Hypoxia-inducible factors regulate human and rat cystathionine
β-synthase gene expression. Biochem J. 458:203–211. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang M, Guo Z and Wang S: Regulation of
cystathionine γ-lyase in mammalian cells by hypoxia. Biochem Genet.
52:29–37. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li M, Nie L, Hu Y, Yan X, Xue L, Chen L,
Zhou H and Zheng Y: Chronic intermittent hypoxia promotes
expression of 3-mercaptopyruvate sulfurtransferase in adult rat
medulla oblongata. Auton Neurosci. 179:84–89. 2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yamawaki K, Kanda H and Shimazaki R: Nrf2
activator for the treatment of kidney diseases. Toxicol Appl
Pharmacol. 360:30–37. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wu QQ, Wang Y, Senitko M, Meyer C, Wigley
WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, et al:
Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases
expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol
Renal Physiol. 300:F1180–F1192. 2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu M, Reddy NM, Higbee EM, Potteti HR,
Noel S, Racusen L, Kensler TW, Sporn MB, Reddy SP and Rabb H: The
Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys
from ischemia-reperfusion injury in mice. Kidney Int. 85:134–141.
2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Han P, Qin Z, Tang J, Xu Z, Li R, Jiang X,
Yang C, Xing Q, Qi X, Tang M, et al: RTA-408 protects kidney from
ischemia-reperfusion injury in mice via activating Nrf2 and
downstream GSH biosynthesis gene. Oxid Med Cell Longev.
2017(7612182)2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu F, Ni W, Zhang J, Wang G, Li F and Ren
W: Administration of curcumin protects kidney tubules against renal
ischemia-reperfusion injury (RIRI) by modulating nitric oxide (NO)
signaling pathway. Cell Physiol Biochem. 44:401–411.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhao L, Xu L, Tao X, Han X, Yin L, Qi Y
and Peng J: Protective effect of the total flavonoids from rosa
laevigata michx fruit on renal ischemia-reperfusion injury through
suppression of oxidative stress and inflammation. Molecules.
21(E952)2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Brady CA and Attardi LD: p53 at a glance.
J Cell Sci. 123:2527–2532. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Molitoris BA, Dagher PC, Sandoval RM,
Campos SB, Ashush H, Fridman E, Brafman A, Faerman A, Atkinson SJ,
Thompson JD, et al: siRNA targeted to p53 attenuates ischemic and
cisplatin-induced acute kidney injury. J Am Soc Nephrol.
20:1754–1764. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wakabayashi N, Slocum SL, Skoko JJ, Shin S
and Kensler TW: When NRF2 talks, who's listening? Antioxid Redox
Signal. 13:1649–1663. 2010.PubMed/NCBI View Article : Google Scholar
|