Introduction
Brugada syndrome (BrS) is an inherited arrhythmia
disorder that was first described in 1992(1). BrS is characterized by ST-segment
elevation in the right precordial leads V1-V2 ≥2 mm, atypical right
bundle branch block, and a high risk of sudden cardiac death (SCD)
due to polymorphic ventricular tachycardia (2). In the majority of cases, the disease
manifests around the age of 35-45 years, but life-threatening
rhythm disturbances may occur at almost any age (3). The prevalence of BrS is ~10:100,000 in
all ethnic groups including Russians (4).
To date, only cardioverter-defibrillator
implantation has proven effectiveness in reducing the risk of SCD
in BrS patients (5-7).
However, the selection of patients for surgical treatment is
difficult, as the prediction of clinical manifestation and SCD risk
assessment remain controversial and need further clarification.
Mutations in at least 23 genes are known to be
responsible for BrS (4). Mutations
in the SCN5A gene are observed in 15-30% of patients
(8), whereas other known genetic
factors account for 5-10% of all cases. It is generally accepted
that BrS is a monogenic autosomal dominant disorder (1,9,10). However, it has been suggested that
the inheritance patterns of BrS do not always fit into classic
Mendelian genetics. It is likely that other genetic and
environmental factors are associated with the manifestation of BrS
(10,11).
There have been two studies highlighting the
possible role of mitochondrial DNA (mtDNA) polymorphisms in the
clinical symptoms of BrS. In the first study, three likely unique
pathogenic heteroplasmic mutations were identified in Iranian
patients with BrS (12). The second
study, performed in Italian BrS patients, showed that mtDNA
haplogroups T and J were found in all symptomatic BrS patients with
a spontaneous type 1 ECG pattern (13). The authors proposed that mtDNA
haplogroups T and J represent important modifying factors for
manifestation of BrS.
The aim of the present study was to assess the
findings of Stocchi et al (13), which showed an association between
BrS and mtDNA haplogroups T and J, and specifically combinations of
mtDNA m.T4216C, m.A11251G, m.C15452A and m.T16126C variants, in a
Russian cohort of patients with BrS.
Materials and methods
Cohort characteristics
A total of 47 index patients (40 males, 7 females)
diagnosed with BrS were included in the present study (median age,
32 years; range, 4-63 years). The male-to-female ratio was 6:1. A
total of 26 patients (55%) had a lone Brugada ECG pattern as an
accidental finding, and 21 patients were symptomatic, with
palpitation/syncope/cardiac arrest and a BrS pattern in their ECG
that was accompanied by concomitant arrhythmias.
Clinical investigation
The present study was performed in accordance with
the principles of the Declaration of Helsinki and the local Ethics
Committee of Petrovsky National Research Centre of Surgery (Moscow,
Russia). Written informed consent was obtained from all individual
participants included in the study. Data obtained from each
individual in the study included personal and familial medical
history, general examination, 12-lead resting ECG, 24-h Holter
monitoring, transthoracic echocardiogram and a pharmacological
challenge test with class Iс anti-arrhythmic drugs. The diagnosis
of BrS was established based on the currently recommended
diagnostic criteria (14).
Genetic screening
Genetic screening of the target mini-panel genes
(SCN5A, SCN1B, SCN2B, SCN3B and SCN4B), encoding the
α- and β-subunits of the Nav1.5 sodium channel, was
performed using semi-conductive sequencing on the IonTorrent
PGM® instrument (Thermo Fisher Scientific, Inc.).
Confirmation of all variants was performed using Next-Generation
Sequencing, resequencing of the regions with low coverage and
cascade familial screening was performed by bidirectional capillary
Sanger Sequencing. Sequencing data were analyzed using the Torrent
Suite software v5.0.5 (Thermo Fisher Scientific, Inc.), Ion
Reporter annotation software v5.12 (Thermo Fisher Scientific, Inc.)
and the Integrative Genomic Viewer visualization tool v2.8.2 (Broad
Institute of MIT). Interpretation of clinically relevant findings
was performed using population databases (ExAC, gnomAD, 1000
Genomes), variant effect predictors (SIFT v1.1.3, PolyPhen2 v2,
MutationTaster, Provean v1.1.3) and splicing analysis tools (UMD
HSF v3.1; NetGene2). Following interpretation, variants were
classified according to American College of Medical Genetics
consensus recommendations (2015) (15). Variants classified as ‘pathogenic’
and ‘likely pathogenic’ (Class V and Class IV) were included in the
final report.
The following reagents were used for hot-start PCR:
HS Taq Turbo buffer, dNTPs (10 mM), Mg2+ (50 mM),
H2O (MQ), HS-Taq polymerase and the corresponding
primers for each exon. All the reagents used were purchased from
Evrogen. Hot-start PCR was performed using a Veriti™ 96-Well
thermal cycler. The thermocycling conditions used were: preheating
at 95˚C for 8 min; followed by 40 cycles of denaturation at 95˚C
for 30 sec, annealing at 60˚C for 30 sec and elongation at 72˚C for
40 sec; and a final elongation step at 72˚C for 1 min.
MtDNA haplogroups were assigned based on the first
hypervariable segment (HVS1) haplotype data obtained by Sanger
Sequencing. A section of the mtDNA D-loop was PCR-amplified using
primers L15997 (5'-CTCCACCATTAGCACCCAAAGC-3') and H408
(5'-CTGTTAAAAGTGCATACCGCCA-3'). The PCR products were then
sequenced on an ABI 3730 DNA Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) using the primers L15997 and Н16526
(5'-AACGTGTGGGCTATTTAGGC-3').
Sanger Sequencing of the PCR products was performed
with BigDye™ Terminator v3.1 Cycle Sequencing kit (Thermo Fisher
Scientific), according to the manufacturer protocol. Primers L15997
and Н16526 (5'-AACGTGTGGGCTATTTAGGC-3') were used as sequencing
primers.
The sequences were aligned to a revised human mtDNA
reference sequence (rCRS) (16).
Each individual haplotype was represented as a list of
substitutions compared with the rCRS. Mitochondrial haplogroup
affiliations were determined using marker HVS1 substitutions,
according to the human mtDNA phylogeny available online (17).
Statistical analysis
The frequencies of the haplogroups in healthy
Russian individuals were based on data from (17). Haplogroup and genotype frequencies
were compared using a χ2 test with Yates correction or a
Fisher's exact test (when the number in any group was <5).
Two-sided 95% confidence intervals for the frequency values were
calculated using the Wilson score method. Statistical analysis was
performed using Statistica v13.2 (Dell). P<0.05 was considered
to indicate a statistically significant difference.
Results
Genetic study
Genetic screening revealed 7 mutations in the
SCN5A gene in 7 probands (14.8%). No disease-causing
mutations were found in the SCN1B-4B genes, and there were
no significant differences found in the clinical manifestation of
BrS between patients with different mutations, such as rate of
syncope, number of SCD cases, age of manifestation, spectrum of
ventricular and supra-ventricular arrhythmias, or ICD implantation
between SCN5A-positive and SCN5A-negative probands
(data not shown). The frequency of the p.H558R allele in the
SCN5A gene was 14.8% in the entire cohort and its frequency
did not differ between symptomatic and asymptomatic patients.
Mitochondrial haplotyping
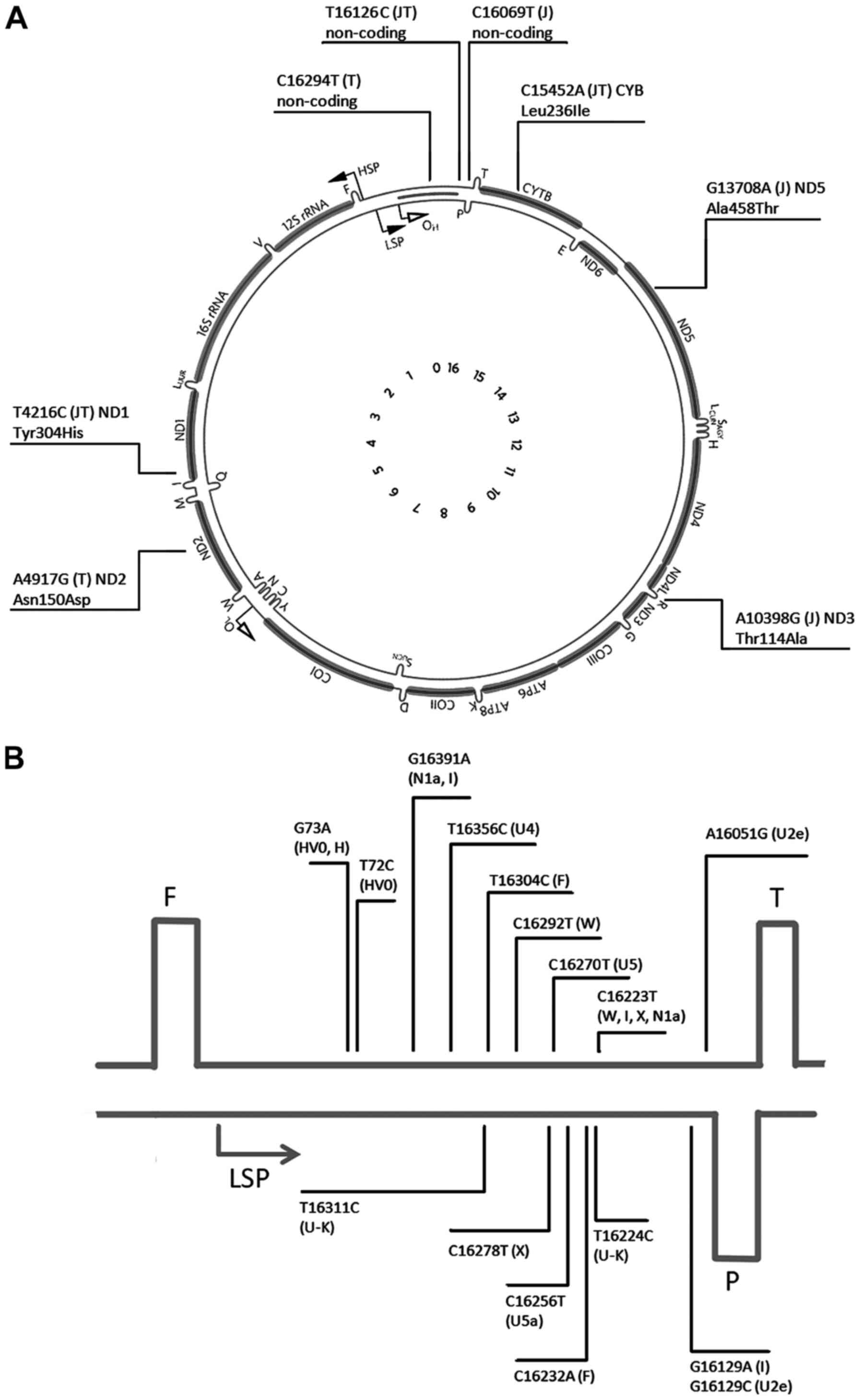
HVS1 in the mtDNA D-loop is known to be the most
variable mtDNA region (Fig. 1)
(18). In the 47 patients with BrS,
37 different HVS1 haplotypes were identified (Table I). The majority of these haplotypes
were only found in 1 sample each, except the ‘CRS’ haplotype
(corresponding to the reference sequence), which was found in 7
individuals, and haplotypes 4, 12 and 22. The mtDNA haplogroups
were then assigned for each individual, using characteristic
substitutions in the HVS1 and substitutions in positions 72 and 73
(data not shown). In total, 11 different mtDNA haplogroups were
identified, five of which (HV, R, X, N1a and F) were only found in
one individual each. The estimated frequencies of the haplogroups
are presented in Table II, along
with 95% confidence intervals. The frequencies of the haplogroup
were compared between patients and healthy Russian individuals
based on data from (17). For only
one haplogroup was a significant difference in the frequency
observed, haplogroup J, which was absent in patients (0%), but was
present at a frequency of 9.7% in the healthy individuals (Fisher's
exact test, P=0.032).
 | Table IMitochondrial DNA first hypervariable
segment haplotypes in the patients with Brugada syndrome. |
Table I
Mitochondrial DNA first hypervariable
segment haplotypes in the patients with Brugada syndrome.
| Number | Variations comparing
to reference mtDNA sequence | Haplogroup | Number of
individuals |
|---|
| 1 | rCRS | H | 7 |
| 2 | C16176T | H | 1 |
| 3 | C16221T | H | 1 |
| 4 | A16293G | H | 2 |
| 5 | C16354T | H | 1 |
| 6 | A16051G, A16162G,
C16266T | H | 1 |
| 7 | A16080G, T16189C,
T16356C | H | 1 |
| 8 | T16092C, T16140C,
A16265G, A16293G, T16311C | H | 1 |
| 9 | A16162G, C16187T | H | 1 |
| 10 | C16188G | H | 1 |
| 11 | A16051G, C16169T | H | 1 |
| 12 | T16298C | HV0 | 3 |
| 13 | G16129A, T16172C,
C16223T, T16311C, G16319A, G16391A | I | 1 |
| 14 | G16129A, T16172C,
C16223T, T16311C, G16391A | I | 1 |
| 15 | T16126C, A16163G,
C16186T, T16189C, C16294T | T (T1a) | 1 |
| 16 | T16126C, A16163G,
C16186T, T16189C, A16293G, C16294T | T (T1a) | 1 |
| 17 | T16126C, A16163G,
C16186T, T16189C, C16294T | T (T1a) | 1 |
| 18 | T16126C, C16294T,
C16296T | T (T2) | 1 |
| 19 | T16126C, C16294T,
C16296T, T16304C | T (T2b) | 1 |
| 20 | T16126C, C16294T,
T16304C | T (T2b) | 1 |
| 21 | A16051G, G16129C,
T16189C, T16362C | U (U2e) | 1 |
| 22 | C16134T,
T16356C | U (U4) | 2 |
| 23 | A16265G, T16356C,
T16362C | U (U4) | 1 |
| 24 | T16136C, T16189C,
C16192T, C16256T, C16270T | U (U5a) | 1 |
| 25 | C16192T, C16256T,
C16270T, C16291T, A16399G | U (U5a) | 1 |
| 26 | C16256T, C16270T,
A16399G | U (U5a) | 1 |
| 27 | T16189C,
C16270T | U (U5b) | 1 |
| 28 | C16192T,
C16270T | U (U5b) | 1 |
| 29 | T16224C,
T16311C | U (K) | 1 |
| 30 | T16224C, T16311C,
G16319A | U (K) | 1 |
| 31 | G16145A, C16223T,
C16292T | W | 1 |
| 32 | C16192T, C16223T,
C16292T, T16311C, T16325C | W | 1 |
| 33 | T16189C, C16223T,
G16255A, C16278T | X | 1 |
| 34 | C16193T | R | 1 |
| 35 | C16168T, C16192T,
A16220C, T16304C | HV | 1 |
| 36 | C16223T,
G16391A | N1a | 1 |
| 37 | T16189C, C16232A,
T16249C, T16304C, T16311C, C16360T | F | 1 |
| Table IIPrevalence of the mtDNA haplogroups
in Russian BrS patients and in general population from the European
region of Russia. |
Table II
Prevalence of the mtDNA haplogroups
in Russian BrS patients and in general population from the European
region of Russia.
| | BrS patients,
n=47 | Controls,
n=953 | |
|---|
| mtDNA
haplogroup | n | Percentage
(CI) | n | Percentage
(CI) | P-value |
|---|
| H | 18 | 38.3
(25.8-52.6) | 386 | 40.5
(37.4-43.7) | 0.882 |
| U (incl. K) | 11 | 23.4
(13.6-37.2) | 240 | 25.2
(22.5-28.0) | 0.919 |
| T | 6 | 12.8
(5.6-25.2) | 85 | 8.9 (7.3-10.9) | 0.525 |
| J | 0 | 0 (0-7.6) | 92 | 9.7 (7.9-11.7) | 0.032a |
| I | 2 | 4.3 (1.5-18.1) | 25 | 2.6 (1.8-3.8) | 0.633 |
| W | 2 | 4.3 (1.5-18.1) | 19 | 2.0 (1.3-3.1) | 0.259 |
| HV0 | 3 | 6.4 (2.2-17.2) | 29 | 3.0 (2.1-4.3) | 0.396 |
| Other | 5 | 10.6
(4.6-22.6) | 77 | 8.1 (6.5-10.0) | 0.581 |
Discussion
Brugada Syndrome is characterized by considerable
genetic and clinical heterogeneity, with incomplete penetrance of
the disease-causing mutations. Generally, it is very difficult to
assess the risk of sudden cardiac death in patients, even if the
causal mutation has been identified (6). Therefore, identification of biomarkers
which will facilitate prediction of the clinical manifestation in
patients with BrS is required. Common genetic variants in other
genes including mitochondrial genes, may be very promising
‘modifying’ targets. MtDNA encodes 13 subunits of the respiratory
chain complex, which are crucial for oxidative phosphorylation
(19). Several mitochondrial
diseases are characterized predominantly by cardiac involvement
including progressive rhythm and conduction disturbances. The best
known example is Kerns-Sayre syndrome, which is caused by a large
mtDNA deletion, and manifests as a progressive AV block and a high
risk of SCD (20). Arrhythmias and
SCD may also occur in patients with mitochondrial encephalopathy,
lactic acidosis and stroke-like episodes syndrome, which is most
frequently caused by the point mtDNA mutation, m.A2343G (21). There have been several studies
showing that common mtDNA polymorphisms may also be modifying
factors in cardiovascular disease phenotypes.
Studies have suggested that mitochondrial
dysfunction (decreased ATP production and increased reactive oxygen
species production) may have an arrhythmogenic effect on the
myocardium (22,23). Decreasing the ATP/ADT ratio results
in the opening of potassium channels, which causes retardation of
impulse conduction (24).
Furthermore, oxidative stress influences the excitability of
cardiomyocytes, partially due to decreased sodium and potassium
entry into cells (25,26). Thus, mitochondrial dysfunction may
reproduce the effects of disease-causing mutations in BrS, and
variations in mitochondrial genome may be promising candidates for
facilitating diagnosis of BrS.
Human mtDNA is highly polymorphic. As mtDNA has no
meiotic recombination, consequent mutations constitute
non-disturbed haplotypes. Related haplotypes are organized into
haplogroups. Each haplogroup is designated by a letter code and has
its own set of nucleotide substitutions in comparison with the
reference (or ancestor) sequence, including amino acid changes and
variants in mitochondrial rRNA and tRNA genes. It is hypothesized
that these haplotypes may have some minor relevance to respiratory
chain function (19).
The first study to show the association between
mtDNA polymorphisms and specific BrS manifestations was performed
by Stocchi et al (13) in
Italian patients. It was found that all symptomatic and half of the
asymptomatic patients with a spontaneous type 1 ECG had mtDNA
belonging to either J or T haplogroups (13). These two mtDNA haplogroups constitute
the phylogenetic cluster JT, defined by the combination of T4216C,
A11251G, C15452A and T16126C substitutions, with T4216C resulting
in amino acid substitution, Tyr304His, in the ND1 subunit and
C15452A resulting in another amino acid substitution, Leu236Ile, in
the Cyt b subunit. In addition, each of the haplogroups has other
polymorphisms which result in amino acid substitutions in different
complexes of the mitochondrial respiratory chain. It is
hypothesized that these missense single nucleotide polymorphisms
together result in reduced oxidative phosphorylation efficiency,
thereby explaining the association with severe forms of BrS.
Stocchi et al (13) found a
high frequency (56%) of the JT cluster in a group of 40 patients,
compared with the frequency in the normal Italian population (20%)
(13).
In the present study, mtDNA polymorphisms in Russian
patients with BrS were studied and it was shown that while the
frequency of the JT haplogroup cluster in the common Russian
population did not differ from its frequency in Italians (~20%), in
Russian patients, haplogroup J was virtually absent, whereas
haplogroup T was observed at almost the same frequency in patients
as it was in the normal population. For the haplogroup J, the
difference was statistically significant. According to the results,
haplogroup J may exhibit a more protective effect than a pathogenic
effect on the development of BrS.
Haplotype definition in Stocchi et al
(13) was based on analysis of the
entire mitochondrial genome with assumption that all mtDNA
haplogroups are the branches of the common phylogenetic tree and
can be unequivocally defined by characteristic combinations of
nucleotide substitutions, as a result of the non-recombinant manner
of mtDNA inheritance. Stocchi et al (13) discussed the combination of four
polymorphisms (T4216C, A11251G, C15452A and T16126C) as a factor
promoting manifestation of Brugada syndrome, and showed that
combination of T4216C and T16126C defined the JT cluster on the
mtDNA tree. Two other substitutions were also present at the root
of the JT cluster; the mtDNA phylogeny is available online
(phylotree.org/tree/JT.htm). Hence, this
combination unambiguously identifies the JT cluster only, and a
higher frequency of this single nucleotide variant (SNV)
combination means a higher frequency of the entire JT cluster. The
presence of another 11 variants separates the JT haplogroup from
the rCRS, which belongs to the haplogroup H2a2 (not to the
ancestral root). The J haplogroup carries 6 more genetic
variations, and the T haplogroup contains an additional 10
characteristic SNVs, which makes for a total of >20 variants
difference (21 for J haplogroup and 25 for the T haplogroup). This
difference reflects the phylogenetic distance of the aforementioned
haplogroups from the reference sequence, but does not correspond to
any specific clinical significance. As most of the mtDNA variants
can be imputed from the haplogroup identification, whole mtDNA
sequencing is redundant for the purpose of the present study.
One possible explanation for the discrepancy between
the present study and Stocchi et al (13) may be that the ‘rough’ classification
of individual haplotypes of mtDNA into major haplogroups does not
reflect geographical differences in mtDNA polymorphisms. Major
mitochondrial haplogroups consist of several sub-haplogroups
defined by additional variants, which can be region-specific
(27). These specific variants may
contribute to the contradictory results. For example, in the
present study, 6 haplotypes belonging to haplogroup T were
represented by three individuals with T1a, one individual with the
T2 root haplotype and two with T2b, whereas Italian patients only
had representatives of sub-haplogroup T2c (13). Similarly, different J sub-haplogroups
may prevail in Italian and Russian populations. Thus, associations
between mtDNA polymorphisms and disease manifestation may be
population specific.
The small size of the clinical group in the initial
study (40 patients with BrS) may introduce a bias into the initial
results. Thus, the preliminary data from Stocchi et al
(13) required a replication study
in a larger number of patients and in a different ethnic group.
The striking contradiction of the results obtained
in the two independent groups of patients with different ethnic
backgrounds suggests that there is no reliable basis for using
mtDNA haplogroups in medical practice for risk stratification of
patients with BrS.
The primary goal of the present study was to assess
the role of common combinations of SNVs (such as haplogroups) in
the manifestation of BrS. Whilst certain individuals may carry
recently occurred unique genetic variants, which may impart some
effect on the clinical course of the disease, the role of these
rare and unique mtDNA variants requires additional methodology and
thus should be the subject of a separate study.
The results of the present study did not confirm the
results of a previously reported association of mtDNA haplogroups J
and T (polymorphism m.T4216C) with BrS or with the more severe form
of the disease (13). In contrast,
haplogroup J was absent in the patients in the present study, but
was present in the normal population at a frequency of 9.7%. Thus,
it seems unlikely that mitochondrial haplogroup J and T contribute
to the clinical manifestation of BrS in Russian patients. However,
clinically relevant biomarkers of manifestation and prognosis of
BrS (electrocardiographical, biochemical and genetic) are required.
Large-scale genome sequencing may provide novel insights and result
in the identification of genes responsible for modification of
clinical phenotypes in patients with BrS.
Acknowledgements
Not applicable.
Funding
Funding
The present study was funded by the Program for
Basic Research of the Russian Academy of Sciences with additional
partial support from RFBR (grant no. 19-04-01322).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MG conceived and designed the study, performed the
analysis, as well as drafted and revised the manuscript. VM
performed the PCR and genotyping, and prepared the illustrations.
VR drafted and revised the manuscript, as well as analyzed the
data. AS performed the genotyping of BrS patients, analyzed the
data and drafted the manuscript. EZ collected the patient data,
performed genetic counseling and ECG analysis, as well as wrote and
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was performed in accordance with the 1964
Helsinki declaration. The head of local Ethics Committee of
Petrovsky National Research Centre of Surgery (Moscow, Russia)
signed a permit to conduct research on Brugada syndrome on
27/09/2019. Written informed consent was obtained from all
individual participants included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brugada P and Brugada J: Right bundle
branch block, persistent ST segment elevation and sudden cardiac
death: A distinct clinical and electrocardiographic syndrome. A
multicenter report. J Am Coll Cardiol. 20:1391–1396.
1992.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Baranchuk A: Brugada Phenocopy. 1st
Edition. The art of recognizing the Brugada ECG. Pattern Imprint:
Academic Press. Published Date: pp154, 27th March 2018.
|
|
3
|
Antzelevitch C and Patocskai B: Brugada
syndrome: Clinical, genetic, molecular, cellular, and ionic
aspects. Curr Probl Cardiol. 41:7–57. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gourraud JB, Barc J, Thollet A, Le
Scouarnec S, Le Marec H, Schott JJ, Redon R and Probst V: The
Brugada syndrome: A rare arrhythmia disorder with complex
inheritance. Front Cardiovasc Med. 3(9)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Priori SG, Wilde AA, Horie M, Cho Y, Behr
ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al:
Executive summary: HRS/EHRA/APHRS expert consensus statement on the
diagnosis and management of patients with inherited primary
arrhythmia syndromes. Europace. 15:1389–1406. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brugada J, Brugada R and Brugada P:
Pharmacological and device approach to therapy of inherited cardiac
diseases associated with cardiac arrhythmias and sudden death. J
Electrocardiol. 33 (Suppl):S41–S47. 2000.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Brugada P, Brugada R, Brugada J and Geelen
P: Use of the prophylactic implantable cardioverter defibrillator
for patients with normal hearts. Am J Cardiol. 83:98D–100D.
1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rattanawong P, Ngarmukos T, Chung EH,
Vutthikraivit W, Putthapiban P, Sukhumthammarat W, Vathesatogkit P
and Sritara P: Prevalence of Brugada ECG pattern in Thailand from a
population-based cohort study. J Am Coll Cardiol. 69:1355–1356.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Juang JM and Huang SK: Brugada syndrome-an
underrecognized electrical disease in patients with sudden cardiac
death. Cardiology. 101:157–169. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shimizu W, Matsuo K, Kokubo Y, Satomi K,
Kurita T, Noda T, Nagaya N, Suyama K, Aihara N, Kamakura S, et al:
Sex hormone and gender difference-role of testosterone on male
predominance in Brugada syndrome. J Cardiovasc Electrophysiol.
18:415–421. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Campuzano O, Brugada R and Iglesias A:
Genetics of Brugada syndrome. Curr Opin Cardiol. 25:210–215.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tafti MF, Khatami M, Rezaei S, Heidari MM
and Hadadzadeh M: Novel and heteroplasmic mutations in
mitochondrial tRNA genes in Brugada syndrome. Cardiol J.
25:113–119. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Stocchi L, Polidori E, Potenza L, Rocchi
MB, Calcabrini C, Busacca P, Capalbo M, Potenza D, Amati F, Mango
R, et al: Mutational analysis of mitochondrial DNA in Brugada
syndrome. Cardiovasc Pathol. 25:47–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bayés de Luna A, Brugada J, Baranchuk A,
Borggrefe M, Breithardt G, Goldwasser D, Lambiase P, Riera AP,
Garcia-Niebla J, Pastore C, et al: Current electrocardiographic
criteria for diagnosis of Brugada pattern: A consensus report. J
Electrocardiol. 45:433–442. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Associationfor Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23(147)1999.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Morozova I, Evsyukov A, Kon'kov A,
Grosheva A, Zhukova O and Rychkov S: Russian ethnic history
inferred from mitochondrial DNA diversity. Am J Phys Anthropol.
147:341–351. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Greenberg BD, Newbold JE and Sugino A:
Intraspecific nucleotide sequence variability surrounding the
origin of replication in human mitochondrial DNA. Gene. 21:33–49.
1983.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wallace DC: The mitochondrial genome in
human adaptive radiation and disease: On the road to therapeutics
and performance enhancement. Gene. 354:169–180. 2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Trivedi M, Goldstein A and Arora G:
Prophylactic pacemaker placement at first signs of conduction
disease in Kearns-Sayre syndrome. Cardiol Young. 28:1487–1488.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ng YS, Grady JP, Lax NZ, Bourke JP, Alston
CL, Hardy SA, Falkous G, Schaefer AG, Radunovic A, Mohiddin SA, et
al: Sudden adult death syndrome in m.3243A>G-related
mitochondrial disease: An unrecognized clinical entity in young,
asymptomatic adults. Eur Heart J. 37:2552–2559. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Montaigne D, Maréchal X, Lacroix D and
Staels B: From cardiac mitochondrial dysfunction to clinical
arrhythmias. Int J Cardiol. 184:597–599. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tse G, Yan BP, Chan YW, Tian XY and Huang
Y: Reactive oxygen species, endoplasmic reticulum stress and
mitochondrial dysfunction: The link with cardiac arrhythmogenesis.
Front Physiol. 7(313)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang KC, Bonini MG and Dudley SC Jr:
Mitochondria and arrhythmias. Free Radic Biol Med. 71:351–361.
2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhou L, Solhjoo S, Millare B, Plank G,
Abraham MR, Cortassa S, Trayanova N and O'Rourke B: Effects of
regional mitochondrial depolarization on electrical propagation:
Implications for arrhythmogenesis. Circ Arrhythm Electrophysiol.
7:143–151. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu M, Liu H and Dudley SC Jr: Reactive
oxygen species originating from mitochondria regulate the cardiac
sodium channel. Circ Res. 107:967–974. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Achilli A, Rengo C, Magri C, Battaglia V,
Olivieri A, Scozzari R, Cruciani F, Zeviani M, Briem E, Carelli V,
et al: The molecular dissection of mtDNA haplogroup H confirms that
the Franco-Cantabrian glacial refuge was a major source for the
European gene pool. Am J Hum Genet. 75:910–918. 2004.PubMed/NCBI View
Article : Google Scholar
|