Introduction
FMS-like tyrosine kinase 3 (FLT3) belongs to the
Receptor Tyrosine Kinase subclass III family, which serves a vital
role in differentiation, proliferation and apoptosis of myeloid
cells (1). The most frequently
observed FLT3 mutations are internal tandem duplications
(FLT3/ITD) in the juxtamembrane domain, which occur in
15-35% of patients with acute myeloid leukemia (AML) (2), and mutations in the tyrosine kinase
activation loop are observed in 5-10% of AML patients (3). Patients with AML with FLT3-ITD
have higher relapse rates (4) and
consequently less favorable disease-free and overall survival rates
(5), particularly in AMLs with a
larger ITD sizes (6), higher allelic
burden (7) or multiple ITDs
(8). Therefore, inhibition of FLT3
has become a potential therapeutic choice, and clinical trials of
inhibitors of FLT3 in AML have been going on for a decade (9). To date, there have been >20 small
molecule inhibitors against FLT3 which have been investigated; some
of which have been examined in clinical trials (10). These include midostaurin (PKC412),
sorafenib (BAY 43-9006), sunitinib (SU11248), tandutinib (MLN518),
lestaurtinib (CEP-701), KW-2449, AKN-032, AC220, ABT-869 and
Quizartinib (AC220) (11,12). The majority of these inhibitors are
structurally heterocyclic compounds that inhibit FLT3 activity by
competing with adenosine triphosphate (ATP) to bind to the tyrosine
kinase domain ATP-binding pocket (13). Functionally, these inhibitors may be
general multikinase inhibitors. Their clinical activities appear to
be mediated by FLT3 inhibition, so their activity is restrained to
AML carrying FLT3-ITDs, and associated with the inhibition of FLT3
phosphorylation and its downstream signaling effectors (14).
Patients diagnosed with acute promyelocytic leukemia
(a subtype of AML) are treated with Vesanoid® [all-trans
retinoic acid (ATRA)]. ATRA promotes the maturation and
differentiation of leukemia cells and is therefore capable of
reducing the symptoms of leukemia by preventing aggregation of
myeloid cells (15). Furthermore,
ATRA has been shown to arrest cell growth, induce cell
differentiation and induce cell death of various types of cancer
cells in vitro (16).
Nonetheless, the clinical applications of ATRA are limited by its
side effects, including acute retinoid resistance,
hypertriglyceridemia, mucocutaneous dryness, nausea, brief recovery
time relapse and drug resistance (17). Additionally, due to its low plasma
concentrations, its medical applications are further reduced.
Therefore, combinations of ATRA and other anticancer drugs were
investigated to overcome these limitations (18). A previous study showed that ATRA can
increase the cytotoxic effects of protein kinase C 412 in AML cell
populations with genetic FLT3 abnormalities (19).
Green tea (from Camellia sinensis) has been
utilized as a Traditional Chinese Medicine for millennia. The
primary active polyphenolic compounds of green tea are catechins
[epicatechin, epigallocatechin and (-)-epigallocatechin-3-gallate
(EGCG)]. Among these catechins, EGCG is the foremost viable
catechin that can reduce the proliferation of cells and induce
apoptosis in cancer cells (20). It
has been shown that EGCG inhibits cancer growth, including lung
(21), prostate (22), colon (23), skin (24) and breast cancer (25).
In previous reports, EGCG (26) and ATRA (19) demonstrated an anti-proliferative
effect on AML cells with FLT3 mutations. In the present study, an
in vitro investigation was performed to assess the effect of
the combination of EGCG and ATRA on FLT3-mutated cell
lines.
Materials and methods
Cell lines and cell culture
Experiments were performed using four human leukemia
cell lines: MOLM-14, MOLM-13 KOCL-48 and MV4-11(26). These above cells were grown in
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
heat-inactivated FBS (Thermo Fisher Scientific, Inc.), 100 IU/ml
penicillin and 0.1 mg/ml streptomycin (cat. no. P4333;
Sigma-Aldrich; Merck KGaA) in a humidified incubator with 5%
CO2 at 37˚C.
Reagents
EGCG (>97% purified powder) was generously gifted
by Dr Yukihiko Hara (Tea Solutions, Hara Office Inc.) and ATRA was
purchased from FUJIFILM Wako Pure Chemical Corporation. The
reagents were dissolved in DMSO. Control cells were cultured with
an equivalent concentration of DMSO as the maximum reagent dose.
Throughout all the experiments, DMSO concentration did not exceed
0.1%, and thus should have had any effect on cytotoxicity (27).
Cell proliferation assay
Cell proliferation assays were performed using a
trypan blue dye exclusion assay as described previously (26,28).
Isobologram
The dose-response interaction between ATRA and EGCG
in the four cell lines were evaluated at the IC50 doses
using an isobologram of Steel and Peckham as described previously
(19,29,30).
Statistical analysis
Data for isobologram were analyzed as described
previously (26,31). The observed data were compared with
the predicted minimum and maximum values for the combined effect.
If minimum predicted value ≤ observed data ≤ maximum predicted
data, the combined effect was additive. However if the mean of the
observed data was higher than the maximum predicted data or lower
than the minimum data, the combined effect was considered
synergistic or antagonistic, respectively. To compare the three
groups (observed, predicted minimum and predicted maximum data), a
Friedman tests followed by a post hoc Nemenyi comparisons test was
used.
To determine whether antagonism or synergism truly
existed, a Wilcoxon signed-ranks test was used to compare the
observed data with the predicted maximum or minimum data for an
additive effect; the data were not normally distributed. P<0.05
suggested the combined effect was considered significant. P≥0.05
suggested the combined the effect was regarded as being additive to
antagonistic or additive to synergistic. Statistical analysis was
performed using R version 4.0.0(32). All experiments were performed at
least three times.
The IC50 values were calculated using
linear approximation of the percentage of survival vs. the
concentration of the drug and was performed using GraphPad Prism
version 8.4.0 (GraphPad Software, Inc.).
Results
ATRA has been shown to suppress cellular
proliferation by inducing apoptosis (19), and EGCG is considered to be an
FLT3-inhibitor which suppresses cell proliferation by disrupting a
FLT3-Hsp90 interaction in FLT3-mutated cell lines (26). The aim of the present study was to
determine whether a combination of the two reagents increased the
effect of these drugs on suppression of cell growth in
FLT3-mutated cell lines. The cytotoxic interaction of two
reagents were examined by isobologram.
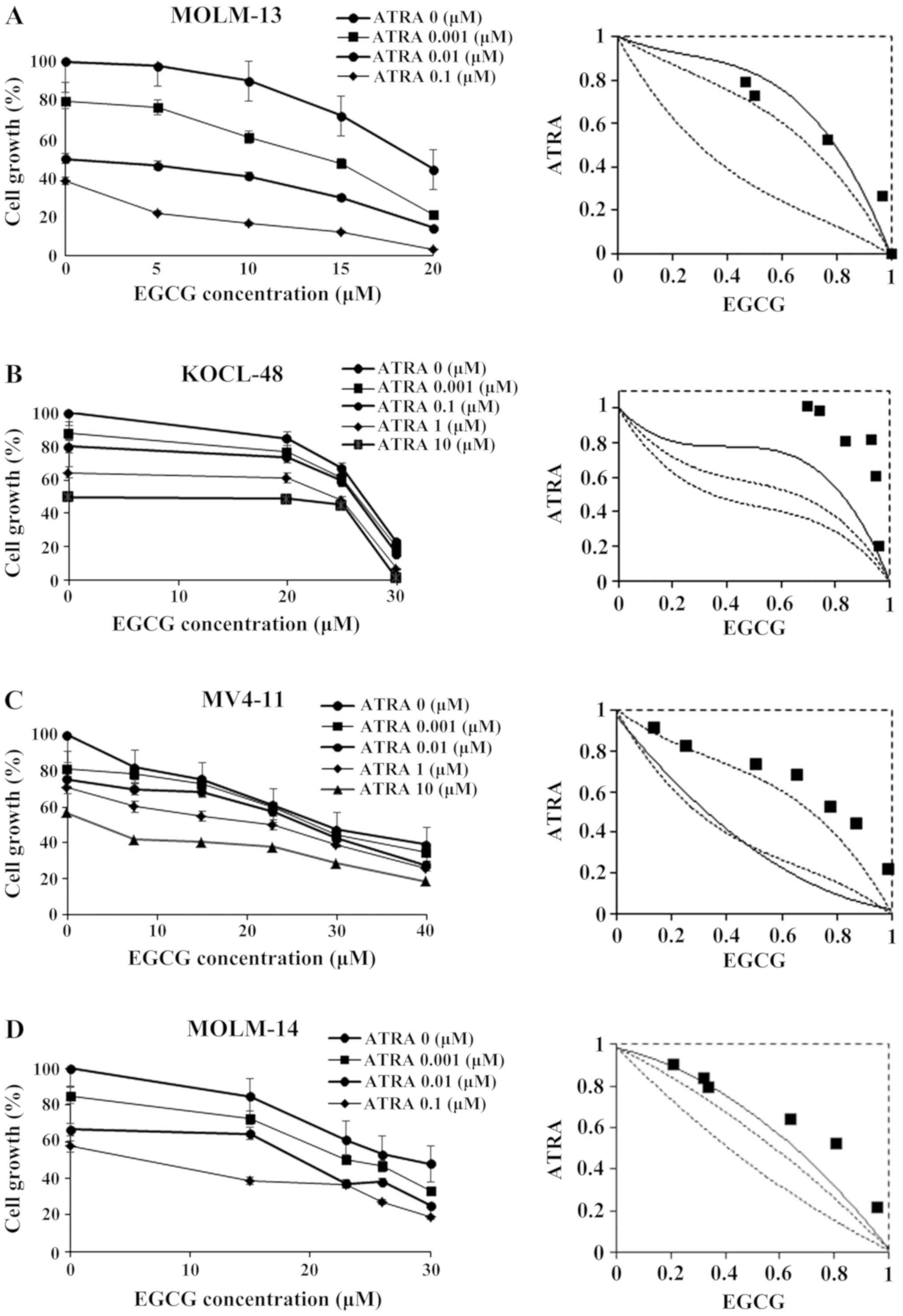
In MOLM-13 cells, one of the data points fell in the
area of sub-additivity (Fig. 1A) but
the results in Table I show that the
mean value of the observed data (0.587) was smaller than that of
the predicted maximum data (0.627) and larger than that of the
predicted minimum data (0.328). Therefore, the combination of EGCG
and ATRA was regarded as additive in MOLM-13 cells.
 | Table IMean values of the observed data and
the estimated minimum and maximum values of combined treatment with
ATRA and EGCG. |
Table I
Mean values of the observed data and
the estimated minimum and maximum values of combined treatment with
ATRA and EGCG.
| | | Predicted values for
the additive effect | | P-valueb | | |
|---|
| Cell line | n | Ob. data | Minimum | Maximum |
P-valuea | Ob./Min | Ob./Max |
P-valuec,
Ob./Max | Effect |
|---|
| MOLM-13 | 5 | 0.587 | 0.328 | 0.627 | | | | | Additive |
| KOCL-48 | 6 | 0.875 | 0.272 | 0.463 | 0.0031 | 0.0026 | 0.1455 | 0.0312 | Antagonism |
| MV4-11 | 7 | 0.676 | 0.301 | 0.593 | 0.0009 | 0.0005 | 0.1472 | 0.0156 | Antagonism |
| MOLM-14 | 6 | 0.69 | 0.43 | 0.616 | 0.0057 | 0.0043 | 0.4804 | 0.0625 | Additive to
antagonistic |
The results showed that the combination of ATRA and
EGCG had an additive cytotoxic effect on MOLM-13 cells compared
with each agent alone. For example, the IC50 of ATRA
alone in MOLM-13 cells was 0.0192±0.0054 µM; however, the
IC50 of ATRA was significantly reduced to 0.0008±0.0008
µM (P<0.01) following combined treatment with EGCG (15 µM)
(Table II).
| Table IIIC50 values of ATRA, EGCG
and ATRA-EGCG combined on leukemia cells. |
Table II
IC50 values of ATRA, EGCG
and ATRA-EGCG combined on leukemia cells.
| Cell line |
|---|
| IC50
value | MOLM-13, µM | MOLM-14, µM | MV4-11, µM | KOCL-48, µM |
|---|
| ATRA | 0.0192±0.0054 | 0.0867±0.0242 | 3.4933±0.2031 | 11.3421±1.0055 |
| EGCG | 18.8333±0.2902 | 26.7251±0.2554 | 26.7531±1.2773 | 26.6055±0.3783 |
| ATRA+EGCG (5
µM) | 0.0088±0.0092 | - | - | - |
| ATRA+EGCG (10
µM) | 0.0031±0.0009 | - | - | - |
| ATRA+EGCG (15
µM) | 0.0008±0.0008 | 0.0259±0.01111 | 0.5683±0.0355 | - |
| ATRA+EGCG (20
µM) | - | - | - | 15.2145±1.0054 |
| ATRA+EGCG (23
µM) | - | 0.0015±0.0027 | 0.0317±0.0630 | - |
| ATRA+EGCG (25
µM) | - | - | - | 0.9861±0.1845 |
| ATRA+EGCG (26
µM) | - | 0.0011±0.0020 | - | - |
The results in Fig.
1B showed that almost all the data points in the KOCL-48 fell
in the area of sub-additivity, suggesting that the combined effect
of ATRA-EGCG was antagonistic, as the mean of the observed data
(0.875) was significantly larger than both the predicted minimum
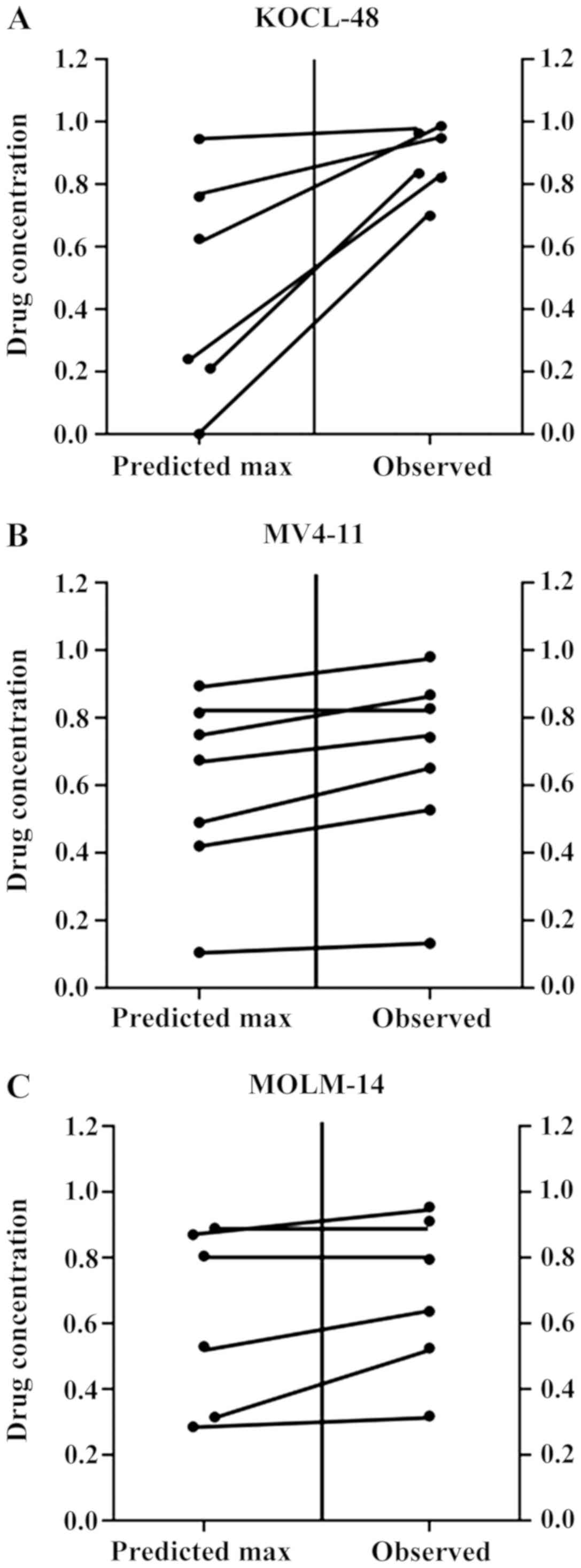
(0.463) and maximum values (0.272) (Table I; P=0.0031). Nemenyi post-hoc tests
were performed, and the results showed there was a significant
difference between the observed data and the predicted minimum data
(P=0.0026), but not between the observed data and the predicted
maximum data (P=0.1455). To determine whether the condition of
antagonism truly existed, a Wilcoxon signed-ranks test was used for
comparing the observed data with the predicted maximum data for an
additive effect (Fig. 2A). The
results showed that the probability value was significant
(P=0.0312) suggesting that the observed data were significantly
higher than the predicted maximum data (Table I), indicative of an antagonistic
effect of simultaneous exposure to the combined treatment in
KOCL-48 cells.
Some data points fell on the border of additivity,
whereas other data points fell in the area of sub-additivity in
MOLM-14 and MV4-11 cells and were thus considered
additive/antagonism (Fig. 1C and
D; MOLM-14, P=0.0057; MV4-11,
P=0.0009). Nemenyi post-hoc test results showed that there were
significant differences between the observed data and the predicted
minimum data (P<0.05), but not between the observed data and the
predicted maximum data in MOLM-14 and MV4-11 (Table I). However, the mean value of the
observed data (MV4-11, 0.676) was significantly higher than the
predicted maximum value (MV4-11, 0.593; P<0.0156; Table I; Fig.
2B) suggesting a true antagonistic effect of the ATRA-EGCG
combination in MV4-11 cells. In contrast, the P-value was 0.0625
suggesting an additive to antagonistic effect in MOLM-14 cells
(Table I; Fig. 2C).
Discussion
ATRA has been used as a major treatment intervention
for patients with APL and functions by inhibiting vascular
endothelial growth factor, which is crucial for angiogenesis
(33). However, the duration of
remission that is induced and maintained by ATRA therapy alone is
short-lived, and ATRA alone fails to induce a second remission in
the majority of patients following relapse (34). In order to address these issues, it
may be necessary to enhance the efficacy of ATRA during the first
treatment regimen. In general, AML is the result of at least two
combined pathophysiological problems, including the acquisition of
chromosomal rearrangements and multiple gene mutations which confer
a proliferative, survival advantage and/or impaired hematopoietic
differentiation (35). Therefore,
administration of a therapy designed to address just one
pathophysiological pathway is likely insufficient for a favorable
response. In addition, administration of anticancer drugs may also
result in severe cytotoxic side effects, restricting the window of
doses which can be administered, thus limiting the potential
efficacy of these therapeutic approaches (36). Through enhancing the effectiveness of
cancer chemotherapy, the use of different combinations of
anticancer drugs may overcome these limitations (36). The majority of anticancer drugs have
distinct molecular mechanisms by which it exert its effects, and
are thus associated with specific cytotoxic side-effects.
Furthermore, for each drug there is an upper limit of concentration
which can be used to achieve effective inhibition of tumor-cell
proliferation whilst minimizing the extent of damage to healthy
cells. A balance of a cocktail of anticancer drugs may therefore
maximize the beneficial effects, reducing the dose of each
individual drug required and thus reducing the associated cytotoxic
side effects of each individual drug (37).
The mechanism of the combined effect of ATRA and
EGCG has only been extensively studied on APL and melanoma. ATRA
enhances the antitumor activity of EGCG by upregulating the
expression of 67-laminin receptor through retinoic acid receptor
(38). EGCG has also been shown to
support ATRA-induced neutrophil differentiation via
death-associated protein kinase 2(39). Another study found that ATRA combined
with EGCG augmented cell differentiation in APL cells by enhancing
the expression of phosphatase and tensin homolog to regulate the
phosphatidylinositol 3-kinase PI3K/Akt/mTOR signaling pathway
(40). However, to the best of our
knowledge, there are no studies reporting on the combined treatment
of ATRA and EGCG in AML cell lines carrying a FLT3 mutation.
Thus, the aim of the present study was to determine the impact of a
combination of ATRA and EGCG on FLT3-mutated AML cell lines.
A limitation of the present study is the fact that APL cell lines
were not used to evaluate the effects of the combined
treatment.
A previous study found that the side effects
associated with ATRA treatment were correlated with the dose given
(17). Therefore, combined treatment
with ATRA and EGCG may maximize the therapeutic efficacy and
mitigate the cytotoxic side effects.
In conclusion, the effects of the combined treatment
with ATRA and EGCG observed in the present study provide
experimental evidence of the potential use of this combination for
treatment of patients with AML who harbor FLT3-mutations.
The novelty of the findings of the present study is that the
combination of ATRA and EGCG resulted in an additive but not
synergistic effect, as seen in APL and melanoma cells. The
underlying mechanism of the combined effect is not understood and
requires further study.
Acknowledgements
We would like to thank Professor Yuko Sato
(University of Tokyo, Tokyo, Japan) for providing the cell lines
used in the present study. We would also like to thank Dr Yukihiko
Hara (Tea Solutions, Hara Office Inc., Tokyo, Japan) for providing
the EGCG powder.
Funding
This study was funded by the Vietnam National
Foundation for Science and Technology Development (grant no.
106.02-2019.50).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
BTKL conceived and designed the study, formulated
the experimental protocols, and prepared the manuscript. HTC
performed the experiments, organized, and analysed the data, and
assisted in the preparation of the manuscript. Both authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Agnes F, Shamoon B, Dina C, Rosnet O,
Birnbaum D and Galibert F: Genomic structure of the downstream part
of the human FLT3 gene: Exon/intron structure conservation among
genes encoding receptor tyrosine kinases (RTK) of subclass III.
Gene. 145:283–288. 1994.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Thiede C, Steudel C, Mohr B, Schaich M,
Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M,
Neubauer A, et al: Analysis of FLT3-activating mutations in 979
patients with acute myelogenous leukemia: Association with FAB
subtypes and identification of subgroups with poor prognosis.
Blood. 99:4326–4335. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Meshinchi S, Stirewalt DL, Alonzo TA,
Zhang Q, Sweetser DA, Woods WG, Bernstein ID, Arceci RJ and Radich
JP: Activating mutations of RTK/ras signal transduction pathway in
pediatric acute myeloid leukemia. Blood. 102:1474–1479.
2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kiyoi H, Yanada M and Ozekia K: Clinical
significance of FLT3 in leukemia. Int J Hematol. 82:85–92.
2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sheikhha MH, Awan A, Tobal K and Liu Yin
JA: Prognostic significance of FLT3 ITD and D835 mutations in AML
patients. Hematol J. 4:41–46. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Meshinchi S, Stirewalt DL, Alonzo TA,
Boggon TJ, Gerbing RB, Rocnik JL, Lange BJ, Gilliland DG and Radich
JP: Structural and numerical variation of FLT3/ITD in pediatric
AML. Blood. 111:4930–4933. 2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Santos FP, Jones D, Qiao W, Cortes JE,
Ravandi F, Estey EE, Verma D, Kantarjian H and Borthakur G:
Prognostic value of FLT3 mutations among different cytogenetic
subgroups in acute myeloid leukemia. Cancer. 117:2145–2155.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gale RE, Green C, Allen C, Mead AJ,
Burnett AK, Hills RK and Linch DC: Medical Research Council Adult
Leukaemia Working Party. The impact of FLT3 internal tandem
duplication mutant level, number, size, and interaction with NPM1
mutations in a large cohort of young adult patients with acute
myeloid leukemia. Blood. 111:2776–2784. 2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pemmaraju N, Kantarjian H, Ravandi F and
Cortes J: FLT3 inhibitors in the treatment of acute myeloid
leukemia: The start of an era? Cancer. 117:3293–3304.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Knapper S: The clinical development of
FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig
Drugs. 20:1377–1395. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kindler T, Lipka D and Fischer T: FLT3 as
a therapeutic target in AML: Still challenging after all these
years. Blood. 116:5089–5102. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cortes JE, Khaled S, Martinelli G, Perl
AE, Ganguly S, Russell N, Krämer A, Dombret H, Hogge D, Jonas BA,
et al: Quizartinib versus salvage chemotherapy in relapsed or
refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A
multicentre, randomised, controlled, open-label, phase 3 trial.
Lancet Oncol. 20:984–997. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pratz K and Levis M: Incorporating FLT3
inhibitors into acute myeloid leukemia treatment regimens. Leuk
Lymphoma. 49:852–863. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fischer T, Stone RM, Deangelo DJ, Galinsky
I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, et
al: Phase IIB trial of oral Midostaurin (PKC412), the FMS-like
tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase
inhibitor, in patients with acute myeloid leukemia and high-risk
myelodysplastic syndrome with either wild-type or mutated FLT3. J
Clin Oncol. 28:4339–4345. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lotan R: Suppression of squamous cell
carcinoma growth and differentiation by retinoids. Cancer Res. 54
(7 Suppl)(1987S-1990S)1994.PubMed/NCBI
|
|
16
|
Amos B and Lotan R: Retinoid-sensitive
cells and cell lines. Methods Enzymol. 190:217–225. 1990.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Conley BA, Egorin MJ, Sridhara R, Finley
R, Hemady R, Wu S, Tait NS and Van-Echo DA: Phase I clinical trial
of all-trans-retinoic acid with correlation of its pharmacokinetics
and pharmacodynamics. Cancer Chemother Pharmacol. 39:291–299.
1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Karmakar S, Banik NL and Ray SK:
Combination of all-trans retinoic acid and paclitaxel-induced
differentiation and apoptosis in human glioblastoma U87MG
xenografts in nude mice. Cancer. 112:596–607. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chi HT, Ly BT, Vu HA, Sato Y, Dung PC and
Xinh PT: Synergistic effect of alltrans retinoic acid in
combination with protein kinase C 412 in FMS-like tyrosine kinase
3-mutated acute myeloid leukemia cells. Mol Med Rep. 11:3969–3975.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Shanafelt TD, Call TG, Zent CS, LaPlant B,
Bowen DA, Roos M, Secreto CR, Ghosh AK, Kabat BF, Lee MJ, et al:
Phase I trial of daily oral Polyphenon E in patients with
asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J Clin
Oncol. 27:3808–3814. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu Q, Qian Y, Chen F, Chen X, Chen Z and
Zheng M: EGCG attenuates pro-inflammatory cytokines and chemokines
production in LPS-stimulated L02 hepatocyte. Acta Biochim Biophys
Sin (Shanghai). 46:31–39. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chuu CP, Chen RY, Kokontis JM, Hiipakka RA
and Liao S: Suppression of androgen receptor signaling and prostate
specific antigen expression by (-)-epigallocatechin-3-gallate in
different progression stages of LNCaP prostate cancer cells. Cancer
Lett. 275:86–92. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sanchez-Tena S, Vizan P, Dudeja PK,
Centelles JJ and Cascante M: Green tea phenolics inhibit
butyrate-induced differentiation of colon cancer cells by
interacting with monocarboxylate transporter 1. Biochim Biophys
Acta. 1832:2264–2270. 2013. View Article : Google Scholar
|
|
24
|
Singh T and Katiyar SK: Green tea
polyphenol, (-)-epigallocatechin-3-gallate, induces toxicity in
human skin cancer cells by targeting β-catenin signaling. Toxicol
Appl Pharmacol. 273:418–424. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Belguise K, Guo S and Sonenshein GE:
Activation of FOXO3a by the green tea polyphenol
epigallocatechin-3-gallate induces estrogen receptor alpha
expression reversing invasive phenotype of breast cancer cells.
Cancer Res. 67:5763–5770. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ly BT, Chi HT, Yamagishi M, Kano Y, Hara
Y, Nakano K, Sato Y and Watanabe T: Inhibition of FLT3 expression
by green tea catechins in FLT3 mutated-AML cells. PLoS One.
8(e66378)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Timm M, Saaby L, Moesby L and Hansen EW:
Considerations regarding use of solvents in in vitro cell based
assays. Cytotechnology. 65:887–894. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Thanh Chi H, Anh Vu H, Iwasaki R, Thao le
B, Hara Y, Taguchi T, Watanabe T and Sato Y: Green tea
(-)-Epigalocatechin-3-gallate inhibits KIT activity and causes
caspase-dependent cell death in gastrointestinal stromal tumor
including imatinib-resistant cells. Cancer Bio Ther. 8:1934–1939.
2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Steel GG and Peckham MJ: Exploitable
mechanisms in combined radiotherapy-chemotherapy: The concept of
additivity. Int J radiat Oncol. 5(85)1979.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kano Y, Ohnuma T, Okano T and Holland JF:
Effects of vincristine in combination with methotrexate and other
antitumor agents in human acute lymphoblastic leukemia cells in
culture. Cancer Res. 48:351–356. 1988.PubMed/NCBI
|
|
31
|
Kano Y, Akutsu M, Tsunoda S, Mori K,
Suzuki K and Adachi KI: In vitro schedule-dependent interaction
between paclitaxel and SN-38 (the active metabolite of irinotecan)
in human carcinoma cell lines. Cancer Chemother Pharmacol.
42:91–98. 1998.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Team RC: R: A language and environment for
statistical computing. Journal. 2018.
|
|
33
|
Kini AR, Peterson LA, Tallman MS and
Lingen MW: Angiogenesis in acute promyelocytic leukemia: Induction
by vascular endothelial growth factor and inhibition by all-trans
retinoic acid. Blood. 97:3919–3924. 2001.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Degos L, Dombret H, Chomienne C, Daniel
MT, Micléa JM, Chastang C, Castaigne S and Fenaux P:
All-trans-retinoic acid as a differentiating agent in the treatment
of acute promyelocytic leukemia. Blood. 85:2643–2653.
1995.PubMed/NCBI
|
|
35
|
Rubnitz JE, Gibson B and Smith FO: Acute
myeloid leukemia. Hematol Oncol Clin North Am. 24:35–63.
2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Nagai S, Takenaka K, Sonobe M, Wada H and
Tanaka F: Schedule-dependent synergistic effect of pemetrexed
combined with gemcitabine against malignant pleural mesothelioma
and non-small cell lung cancer cell lines. Chemotherapy.
54:166–175. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Torchilin V: Antibody-modified liposomes
for cancer chemotherapy. Expert Opin Drug Deliv. 5:1003–1025.
2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lee JH, Kishikawa M, Kumazoe M, Yamada K
and Tachibana H: Vitamin A enhances antitumor effect of a green tea
polyphenol on melanoma by upregulating the polyphenol sensing
molecule 67-kDa Laminin Receptor. PLoS One.
5(e11051)2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Britschgi A, Simon HU, Tobler A, Fey M and
Tschan M: Epigallocatechin-3-gallate induces cell death in acute
myeloid leukaemia cells and supports all-trans retinoic
acid-induced neutrophil differentiation via death-associated
protein kinase 2. Br J Haematol. 149:55–64. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yao S, Zhong L, Chen M, Zhao Y, Li L, Liu
L, Xu T, Xiao C, Gan L, Shan Z and Liu B:
Epigallocatechin-3-gallate promotes all-trans retinoic acid-induced
maturation of acute promyelocytic leukemia cells via PTEN. Int J
Oncol. 51:899–906. 2017.PubMed/NCBI View Article : Google Scholar
|