Introduction
Acute myeloid leukemia (AML) accounts for ~20% of
all cases of pediatric leukemia (1).
Although the prognosis of pediatric AML has improved in recent
decades, it trails that of other types of pediatric cancer, as
30-40% of children with AML eventually succumb to the disease
(1-3).
The treatment strategies for pediatric AML include intensive
multimodal chemotherapy with or without stem cell transplantation,
and cytarabine and anthracyclines have remained the primary choices
of chemotherapy for >30 years (1,2,4,5). As
clinical outcomes have not improved, even with intensive
contemporary chemotherapeutic regimens and/or stem cell
transplantation, newer targeted drugs are required for
incorporation into treatment plans (5,6).
Recently, various newer targeted therapies have
emerged, most of which target AML cells with specific genetic
alterations (7). Among them, FLT3
inhibitors, such as midostaurin and gilteritinib, and IDH
inhibitors, such as enasidenib and ivosidenib have already been
approved for use in clinical settings (7). These newer drugs selectively target AML
cells with specific features; hence, the genomic characterization
of AML cells is becoming increasingly important in the clinical
setting. Using next-generation sequencing (NGS), several studies
have reported on the value of performing NGS for adult patients
with AML (8-12).
However, there are comparatively fewer reports focusing on
childhood AML, particularly in cases of relapsed AML (13,14).
In the present study, panel-based, targeted NGS for
the molecular characterization of AML cells from pediatric patients
was retrospectively performed. The objective of this study was to
determine whether it was possible to obtain clinically useful
information for children with AML through this NGS approach.
Materials and methods
Patients
A total of 27 children aged 0-18 years who were
diagnosed with AML between January 2000 and December 2017 in
Okayama University Hospital, Kochi Health Sciences Center, St.
Marianna University School of Medicine Hospital or Hokkaido
University Hospital were enrolled in the present study. The ratio
of boys to girls was 17:10 in the present cohort and the median age
at diagnosis was 6 years (range, 0 months to 15 years). The
treatment protocols were diverse, including those from the Japanese
Pediatric Leukemia/Lymphoma Study Group AML-99(15), AML-05(16) and AML-12 studies, and are listed in
Table I. The standard chimeric
fusion gene screening varied has changed over time in Japan. AML-05
protocol included the PCR-based detection of eight frequent
chimeric gene fusions; RUNX1-RUNXT1, CBFB-MYH11,
KMT2A-MLLT3, KMT2A-MLLT4, KMT2A-MLLT1,
FUS-ERG, NUP98-HOXA9 and PML-RARA, and the
AML-12 protocol included eight gene fusions; RUNX1-RUNXT1,
CBFB-MYH11, KMT2A-MLLT3, KMT2A-MLLT4,
BCR-ABL, FUS-ERG, NUP98-NSD1 and PML-RARA. In
our previous study, the clinical courses of UPN 6 and UPN 25 was
reported (17,18). The present study was performed in
accordance with the Declaration of Helsinki and other applicable
guidelines (19-21).
The present study was approved by the Institutional Ethics
Committee of Okayama University Hospital, and informed consent was
obtained the patients and/or their legal guardians.
 | Table IClinical information of the analyzed
patients. |
Table I
Clinical information of the analyzed
patients.
| A, Relapsed
AML |
|---|
| UPN | Age | Sex | Disease
subtype | Chimeric gene | Karyotyping | Initial
therapya | SCT at CR1 | Duration of CR1,
months | Karyotype at
relapse | Outcome | Institute |
|---|
| 1 | 12 years | M | FAB M5 | KMT2A-MLLT3 | 46, XY,
add(11)(q23), der(21)t(1;21) (q11;q11.2) [9/20], 46, sl,
der(19)t(11;19) (q13;p13) [11/20] | AML 05 | No | 24 | 47, XY, add(11)
(q23), +marl | Alive | OU |
| 2 | 4 years | M | FAB M6 (RAEB) | None | 46, XY, inv(9)
(p11q13) | Other | Yes | 7 | 46, XY,
t(2;5)(q33;q31), add(6)(q21), t(6;12)(q13;p13), inv(9)(p11q13) | Alive | OU |
| 3 | 5 months | M | FAB M7
(non-Down-syndrome) | None | 49, XY, +4,
?t(4;5)(q21;q15), del(12)(p?), +19, +22 | AML 12 | No | 13 | 50, XY, +4,
?t(4;5)(q21;q15), +8, del(12)(p?), +19, +22 | Alive | OU |
| 4 | 10 years | M | FAB M5 | None | 48, XY, +16,
+19 | AML 05 | No | 9 | 46, XY,
add(7)(q32), ?t(10;11)(p12;q23) | Succumbed to
disease | OU |
| 5 | 4 years | F | FAB M7
(non-Down-syndrome) | None | 46, XX, add(5)
(p13), del(6)(q), add(12)(p11.2) | Other | No | 22 | 46, XX,
add(5)(p13), del(6)(q), add(12)(p11.2) | Alive | OU |
| 6 | 12 years | F | FAB M2 | None | 46, XX | AML 05 | Yes | 7 | NA | Succumbed to
disease | KHSC |
| 7 | 7 years | F | FAB M2 | None | 46, XX | AML 99 | No | 10 | 46, XX | Succumbed to
disease | HU |
| 8 | 1 year | M | FAB M7
(Down-syndrome) | None | 47, XY, +21 | Other | No | 12 | 45, XY, -10,
der(17) t(17;18)(p11;q11), -18, +21 | Dead (CR) | HU |
| 9 | 13 years | M | FAB M4 | NA | 46, XY, t(6;11)
(q27;q23) | AML 99 | No | 18 | 46, XY,
t(6;11)(q27;q23) | Succumbed to
disease | HU |
| 10 | 1 year | M | FAB M5b | NA | 47, XY, t(9;11)
(p22;q23), +mar1 | AML 99 | No | 6 | NA | Alive | HU |
| 11 | 14 years | M | FAB M1 | None | 46, XY,
del(9)(q?) | AML 05 | No | 18 | 46, XY | Alive | HU |
| 12 | 11 years | F | T/Myeloid | None | 46, XX, t(1;2)
(p31;p16) | AML 05 | No | 13 | 46, XX,
t(1;2)(p31;p16) | Alive | HU |
| 13 | 5 years | M | FAB M7
(non-Down-syndrome) | None | 47, XY, del(9)
(q12q34), del(12)(p12), +21 | Other | Yes | 19 | 47, XY, +8,
del(9)(q12q34), t(10;17)(q11.2;q25), del(12)(p12) | Succumbed to
disease | HU |
| 14 | 5 years | F | FAB M4 | None | 46, XX, der(2)
t(11;10;2)(q21; q11.2;q37), der(10) add(10)(p11.2) t(11;10;2),
der(11) t(11;10;2) | AML 05 | No | 24 | 46, XX | Succumbed to
disease | HU |
| 15 | 14 years | M | T/Myeloid | None | 47, XY, del(4)(q?),
+22, inc | AML05 | No | 4 | 47,XY, del(4)(q?),
22 | Alive | SMU |
| B, Non-relapsed
AML |
| UPN | Age | Sex | Disease
subtype | Chimeric gene | Karyotyping | Initial
therapya | SCT at CR1 | Duration of CR1,
months | Karyotype at
relapse | Outcome | Institute |
| 16 | 10 years | M | FAB M3 | PML-RARA | 46, XY, t(15;17)
(q22;q11~21) | AML99 | No | - | - | Alive | OU |
| 17 | 9 years | F | FAB M3 | PML-RARA | 46, XX, t(15;17)
(q22;q12) | AML 99 | No | - | - | Alive | OU |
| 18 | 8 months | F | FAB M5a | KMT2A-MLLT10 | 46, XX, t(10;11)
(p12;q23) | AML 12 | Yes | - | - | Alive | OU |
| 19 | 13 years | M | FAB M4Eo | CBFB-MYH11 | 47, XY, +8,
inv(16)(p13.1q22) | AML 05 | No | - | - | Alive | OU |
| 20 | 7 years | M | FAB M2 | RUNX1-RUNXT1 | 45, X, -Y, t(8;21)
(q22;q22) | AML 05 | No | - | - | Alive | OU |
| 21 | 15 years | M | AML with
myelodysplasia-related changes | None | 46, XY | Other | Yes | - | - | Dead (CR) | OU |
| 22 | 6 years | F | FAB M2 | CBFB-MYH11 | 46, XX, inv(16)
(p13.1q22) | AML 05 | No | - | - | Alive | OU |
| 23 | 5 years | F | FAB M2 | RUNX1-RUNXT1 | 46, XX, t(8;21)
(q22;q22) | AML 05 | No | - | - | Alive | OU |
| 24 | 3 years | M | FAB M2 | RUNX1-RUNXT1 | 46, XY, t(1;21;8)
(p36;q22;q22), del(9)(q13q22) | AML 99 | No | - | - | Alive | OU |
| 25 | 0 months | F | FAB M5 | KAT6A-CREBBP |
t(8;16)(p11;p13) | AML 99 | No | - | - | Alive | OU |
| 26 | 11 months | M | FAB M5 | KMT2A-MLLT3 | 46, XY | AML 99 | No | - | - | Alive | OU |
| 27 | 1 year | M | FAB M2 | RUNX1-RUNXT1 | 46, XY,
t(8;21)(q22;q22), del(9)(q?), add(11)(q23) | AML 99 | No | - | - | Alive | OU |
DNA isolation
Somatic DNA was obtained from bone marrow samples at
diagnosis and each episode of relapse, whereas germline DNA was
obtained from a buccal swab or peripheral blood in CR status. DNA
was extracted using a QIAamp DNA Blood Mini kit (Qiagen, Inc.) and
quantified using a NanoDrop 2000 (Thermo Fisher Scientific, Inc.)
and a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific,
Inc.) according to the manufacturers' protocol.
Targeted NGS approach
Targeted sequencing of >150 cancer-related genes
was performed as described previously (22-24).
The targeted gene lists are shown in Table SI and patient allocation is shown in
Table SII. These gene panels were
generated using an online design tool for HaloPlex (Agilent
Technologies, Inc.), and target enrichment was performed using the
HaloPlex standard protocol. Samples were then sequenced using MiSeq
(Illumina, Inc.). Read alignment to the hg19 reference genome was
performed using Burrows-Wheeler Aligner (bio-bwa.sourceforge.net) and variant calling was
performed using SureCall version 3.0 (Agilent Technologies,
Inc.).
Variant prioritization and assessment
of pathogenicity
Synonymous or non-coding variants and single
nucleotide polymorphisms reported with a frequency of >1% in
various databases (dbSNP, 1000gp and Human Genetic Variation
Database) were excluded. Variant bases that had >5 reads in each
sample were used for the next step. Genetic variations that were
constantly detected from diagnostic, remission and relapse samples
with a variant allele frequency (VAF) ≥0.2 were regarded as
candidate germline alterations. Genetic alterations that were
rarely detected, or not detected at all, from remission samples but
were detected from diagnostic and/or relapse samples with a VAF
≥0.05 were regarded as candidate somatic alterations with reference
to a previous study (25). To
exclude the possibility of false-positive findings, differences in
VAF between normal and diagnostic/relapse samples were assessed
using a Fisher's exact test. P<0.01 was considered to indicate a
statistically significant difference. Finally, for germline and
somatic alterations, the read quality was checked using IGV
software version 2.3 (Broad Institute).
Results
Patient characteristics
The clinical information of the analyzed patients is
shown in Table I. All patients were
Japanese. The present cohort included one patient with
Down-syndrome (UPN 8) and no other patients had a known underlying
congenital condition. Remission samples were obtained by buccal
smear from patient No. 2 and the sample collection was performed
during remission. All relapsed patients lost their first remission
within 24 months; 6 out of 15 patients experienced their relapse
within 12 months, which is considered to be an adverse prognostic
factor for survival (26).
Descriptive results from sequencing
runs
The average number of total reads was 1,917,277
(range, 998,341-4,333,332) and the average read length was 116-136.
The read depth in analyzable target regions per sample ranged from
168-757x. In total, 70.49-97.51% of analyzable regions were covered
by at least 20 reads, 66.72-95.24% were covered by at least 50
reads and 58.15-91.05% were covered by at least 100 reads. These
quality metrics data were obtained from analysis using SureCall
version 3.0 software (Agilent Technologies, Inc.) and the details
are shown in Table SII.
The results of NGS are shown in Table II. A total of 26 single nucleotide
variations (SNVs) and insertions/deletions (indels) were identified
at diagnosis, and 22 SNVs and indels at relapse for 15 patients
with relapsed AML, as well as 12 SNVs and indels for the leukemia
samples of 12 patients without relapse.
| Table IIGene alterations detected at
diagnosis and relapse. |
Table II
Gene alterations detected at
diagnosis and relapse.
| A, Relapsed
AML |
|---|
| UPN | Disease
subtype | SNVs at diagnosis
(VAF) | SNVs at relapse
(VAF) |
|---|
| 1 | FAB M5 | KRASp.G12V
(0.296) | None |
| 2 | FAB M6 (RAEB) |
PTPN11p.G60V(0.83) PTPN11
p.V45L (0.10) |
PTPN11p.G60V(0.44) |
| 3 | FAB M7
(non-Down-syndrome) | KRAS p.A146T
(0.09) | None |
| 4 | FAB M5 | None | None |
| 5 | FAB M7
(non-Down-syndrome) | GATA1p.R270W
(0.43) KRAS p.G12A (0.43) IKZF1 p.F154Y
(0.38)a
IKZF1p.L161H (0.38)a |
GATA1p.R270W(0.23) KRAS
p.G12A (0.19) IKZF1p.F154Y(0.17)a
IKZF1p.F161H(0.17)a |
| 6 | FAB M2 | FLT3-ITD
(0.572) | FLT3-ITD
(0.689) |
| 7 | FAB M2 | PTPN11
p.I479F (0.40) + c.1473-1481dela |
PTPN11p.I479F (0.35) +
c.1473-1481dela |
| 8 | FAB M7
(non-Down-syndrome) | GATA1
p.P50fs (0.30) | GATA1
p.P50fs (0.05) |
| 9 | FAB M4 | KRAS p.G12V
(0.41) SMARCA4p.R979Q (0.06)a | MLH1p.A586S
(0.06)a |
| 10 | FAB M5b | KRAS p.G13D
(0.32) |
RUNX1p.Q390fs(0.2) |
| 11 | FAB M1 | GATA2
p.R362Q (0.11) CEBPAp.Q312HR (0.99)a |
CEBPAp.D63fs(0.37)a CEBPAp.Q312HR
(0.39)a |
| 12 | T/Myeloid | JAK3 p.L857P
(0.10) NOTCH1p.V1721E (0.33) STAT5Bp.N642H(0.11)
NF1 p.L2317H (0.05)a | JAK3 p.L857P
(0.13) NOTCH1p.V1721E (0.33) IL7R p.L243GTARCV
(0.10)a VHL
p.L85fs (0.11) |
| 13 | FAB M7
(non-Down-syndrome) | None | None |
| 14 | FAB M4 | U2AF1 p.R35L
(0.53) KRAS p.G12D (0.37) | KRAS p.G12D
(0.23) |
| 15 | T/Myeloid | TP53 p.K164E
(0.998) PHF6 p.R274fs (0.724) NPM1 p.L287fs
(0.579) | TP53 p.K164E
(0.858) PHF6p.R274fs (0.611) NPM1 p.L287fs (0.53)
MPLp.A486V(0.064)a |
| B, Non-relapsed
AML |
| UPN | Disease
subtype | SNVs at diagnosis
(VAF) | SNVs at relapse
(VAF) |
| 16 | FAB M3 | None | - |
| 17 | FAB M3 | None | - |
| 18 | FAB M5a | None | - |
| 19 | FAB M4Eo | KIT p.D816Y
(0.28) | - |
| | | WT1 p.H448Y
(0.22)a | - |
| | | NRAS p.Q61K
(0.08) | - |
| 20 | FAB M2 | JAK3 p.M511I
(0.13) | - |
| 21 | AML with
myelodysplasia-related changes | None | - |
| 22 | FAB M2 | NRAS p.G13D
(0.37) | - |
| | | WT1 p.D447N
(0.34) | - |
| 23 | FAB M2 | NRAS p.Q61K
(0.25) | - |
| 24 | FAB M2 | KRAS p.G12D
(0.52) | - |
| 25 | FAB M5 | None | - |
| 26 | FAB M5 | FLT3 p.D839G
(0.21) | - |
| | | FLT3 p.Y591D
(0.08) | - |
| | | FLT3 p.D839N
(0.07) | - |
| 27 | FAB M2 | KIT p.N822K
(0.43) | - |
Somatic genetic alterations at
diagnosis and AML subtypes
In the present study, seven genes were recurrently
mutated. KRAS was mutated in 7 patients, NRAS was
mutated in 3 patients, and KIT, GATA1, WT1,
PTPN11, JAK3 and FLT3 were each mutated in 2
patients. As previously reported, KIT was mutated in
patients with core-binding factor AML (UPN 19 and UPN 27), and
GATA1 was mutated in patients with acute megakaryoblastic
leukemia with or without Down-syndrome (UPN 5 and UPN 8) (27-29).
The present cohort included 2 patients with mixed-phenotype acute
leukemia, and these patients harbored mutations previously reported
in a larger study (30). Other
mutations were not apparently associated with a specific type of
AML. Most detected mutations have been reported in the Catalogue Of
Somatic Mutations In Cancer database (cancer.sanger.ac.uk/cosmic), but some alterations were
not. These mutations were thought to be variants of unknown
significance and are indicated with a superscripted letter in
Table II.
Prognostic genetic alterations
detected at diagnosis
According to the European LeukemiaNet (ELN)
guideline, several factors are associated with the prognosis of AML
(31). Among these, two high-risk
genetic alterations (FLT3-ITD in patient 6 and TP53
alteration in patient 15) and one low-risk genetic alteration
(CEBPA mutation in patient 11) were added to the known
cytogenetic risk factors (Table
III). According to the guidelines, RUNX1 or ASXL1
alterations are indicated not to be used as adverse prognostic
markers if they are present concurrently with favorable-risk AML
subtypes. However, TP53 alterations are regarded as an
independent adverse prognostic factor, thus UPN 15 was placed in
the ELN high-risk group. The gene alterations which were defined in
the ELN guideline or recurrently detected in the present study are
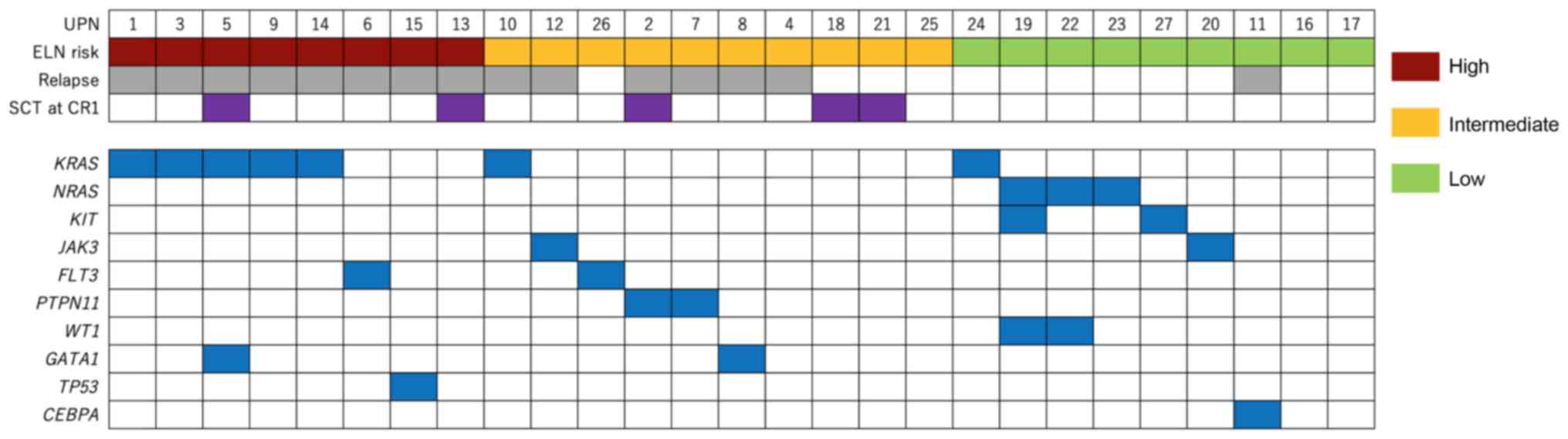
summarized in Fig. 1.
| Table IIIPrognostic genetic alterations at
diagnosis according to the European LeukemiaNet guidelines. |
Table III
Prognostic genetic alterations at
diagnosis according to the European LeukemiaNet guidelines.
| UPN | Low-risk
features | Intermediate risk
features | High-risk
features. |
|---|
| 1 | No |
KMT2A-MLLT3 | Complex
karyotype |
| 2 | No | No | No |
| 3 | No | No | Complex
karyotype |
| 4 | No | No | No |
| 5 | No | No | Complex
karyotype |
| 6 | No | No |
FLT3-ITD |
| 7 | No | No | No |
| 8 | No | No | No |
| 9 | No | No |
t(6;11)(q27;q23) |
| 10 | No |
t(9;11)(p22;q23) | No |
| 11 | CEBPA
p.Q312HR | No | No |
| 12 | No | No | No |
| 13 | No | No | Complex
karyotype |
| 14 | No | No | Complex
karyotype |
| 15 | No | No | TP53
p.K164E |
| 16 |
PML-RARA | No | No |
| 17 |
PML-RARA | No | No |
| 18 | No | No |
KMT2A-MLLT10 |
| 19 |
CBFB-MYH11 | No | No |
| 20 |
RUNX1-RUNXT1 | No | No |
| 21 | No | No | No |
| 22 |
CBFB-MYH11 | No | No |
| 23 |
RUNX1-RUNXT1 | No | No |
| 24 |
RUNX1-RUNXT1 | No | No |
| 25 | No | No | No |
| 26 | No |
KMT2A-MLLT3 | No |
| 27 |
RUNX1-RUNXT1 | No | No |
Mutational changes between diagnosis
and relapse
A total of 15 patients who experienced relapse were
analyzed. Among these, six harbored KRAS mutations at
diagnosis. However, four of the six patients lost these mutations
at relapse. None of the patients gained new RAS pathway mutations
at relapse.
UPN 11 had a CEBPA p.Q312HR insertion-type
alteration at diagnosis, and this alteration appeared to be
homozygous as its VAF was notably high (0.99). At relapse however,
the VAF of CEBPA p.Q312HR alteration decreased, and a new
p.D53fs alteration appeared, leading to a ‘double hit’ status.
Germline variations
In the present study, matched samples at diagnosis,
remission and relapse (if any) among patients with childhood AML
was analyzed, and this approach enabled detection of germline
variations. Although candidates of germline variations were
detected in 8 patients, none were regarded as pathogenic or likely
pathogenic according to published recommendations (19,21).
These candidate genes are listed in Table SIII.
Discussion
In the present study, matched samples obtained from
pediatric patients with AML at diagnosis, remission and relapse (if
any) were analyzed using a panel-based NGS method. Several studies
have reported the utility of NGS for analysis of AML in adult
patients (8-12).
However, there are comparatively fewer reports focusing on
childhood AML, particularly in cases of relapsed AML (13,14).
The utility of NGS should be discussed separately in
adult and pediatric patients. As was shown in the present study,
there are significant differences in genetic alterations between
adult and pediatric patients with AML. Whereas mutations in
epigenetic components or spliceosome complexes are common among
adults with AML (11), these
mutations were notably less common amongst children with AML, based
on the results of the present study. However, large structural
aberrations such as chromosomal translocations are more common
amongst children with AML (11,14). As
a result, disease stratification guidelines or newer drugs
developed for adults with AML may not always be suitable for
children.
The panel-based NGS strategy has been used several
times in our previous studies for hematological malignancies, and
the results of this method have been compared with conventional
approaches, such as the multiplex ligation-dependent probe
amplification or Sanger sequencing in our previous studies
(22-24).
Based on the panel-based NGS approach, several prognostic genetic
alterations for patients with AML were detected in the present
study. The ELN guidelines identified several cytogenetic
alterations and a smaller number of genetic mutations as prognostic
factors. In the present cohort, additional cytogenetic risk factors
possessed prognostic implications than the genetic mutations. Using
this approach, two high-risk genetic mutations were detected
amongst patients who relapsed but did not possess any cytogenetic
risk factors (UPN 6 and UPN 15). Conversely, none of the
non-relapsed patients possessed adverse prognostic genetic
mutations, and non-relapsed patients were enriched in low risk
genetic alternations defined in the ELN guidelines. Thus, it may be
possible to more accurately predict the prognosis of patients with
AML using this approach. However, as noted above, there are
significant differences in genetic alterations between adult and
pediatric patients with AML. Previous larger studies suggested that
current guidelines, including the ELN guidelines, are not adequate
for children with AML (14,32,33),
thus, there is a need for the development of a pediatric-specific
guidelines for more precise stratification.
FLT3-ITD has a prognostic impact in pediatric
AML (34,35), and this alteration was detected using
the panel-based NGS method. However, the clinical impact of
FLT3-ITD has been reported to be modulated by other sequence
aberrations (WT1 mutation, NUP98-NSD1 or NPMI
mutations for adult AML) (14,36). In
this context, panel-based sequencing may be more useful than
conventional approaches that detect only FLT3-ITD. However,
the ELN guidelines recommend the use of DNA fragment analysis to
determine the ratio of FLT3-ITD and prognosis. To confirm
the usefulness of the NGS approach, a direct comparison of standard
procedures and the NGS approach is required.
The number of genes that should be assessed has
increased; however, one large study found that a limited number of
genes are recurrently mutated in pediatric patients with AML
(14); where several genetic
analytical methods, including whole-genome and targeted DNA
sequencing were performed. Mutations in only 5 genes (FLT3,
NPM1, WT1, CEBPA and KIT) were present
in >5% of patients, and <40 gene mutations were reported in
>2% of patients. This previous study illustrated the need to
focus on these 40 genes to detect recurrent gene mutations in AML
cells obtained from pediatric patients, and that panel-based
sequencing is an ideal approach in a clinical setting. Furthermore,
Morita et al (37) reported
that the clearance of somatic mutation at remission was associated
with significantly improved survival and a lower risk of relapse.
This strategy requires a sufficient read depth to detect mutations
with a VAF <1%; contrarily, the approach used in the present
study could not reach that read depth due to low throughput and
relatively high number of targeted genes. Limiting the number of
targeted genes to 40 recurrently mutated genes will increase the
read depth and potentially enable detection of gene alterations
with lower VAFs.
The panel-based approach used in the present study
also offers potentially useful information regarding targetable
genetic alterations. Cytotoxic chemotherapy primarily based on
cytarabine and anthracyclines with or without stem cell
transplantation has long been the mainstay of AML treatment
(1). The curative rate of pediatric
AML steadily improves with increasing doses of these drugs;
however, this leads to substantial treatment-related complications
in vulnerable pediatric populations (1,16,26).
Hence, newer targeted therapies are desired. These targeted drugs
include midostaurin, gilteritinib, enasidenib and ivosidenib, which
target leukemic cells with specific genetic alterations such as
FLT3, KIT, MEK, DOT1L or BET
alterations (5,6); hence, genomic characterization of AML
cells is becoming increasingly important in the clinical setting.
Among these potentially targetable gene alterations, 6 patients had
RAS pathway mutations at the time of AML diagnosis. However, in the
present cohort, a notably high percentage of patients (four out of
six cases, 66.7%) lost the KRAS mutations at relapse. Thus,
RAS pathway mutations should be chosen with caution as treatment
targets.
The present study has several limitations. First,
the study was retrospective, and it was not possible to clarify
whether dose increases in patients who had high-risk features would
improve their prognoses. Second, the method used in the present
study has a disadvantage of comparatively low throughput. Third,
potential false positives were excluded, thus several relevant
genetic alterations may have been missed. To detect minor clones
with low variant frequencies at diagnosis, the number of genes to
be targeted should be limited, as noted above. The genetic events
including TP53 loss of heterozygosity in UPN 15 should have
also been illustrated using other experiments, such as fluorescent
in situ hybridization, but this could not be achieved due to
the sample availability and quality. Relatively small patient
numbers is another disadvantage of the present study. Furthermore,
the ELN guidelines were established for adult patients; hence its
validity in a larger cohort of pediatric patients with AML requires
validation.
In summary, the panel-based targeted sequencing
approach used in the present study may be useful for revealing the
genetic background of pediatric AML, and may facilitate the precise
prediction of patient prognosis and detection of druggable gene
alterations. Incorporating this method into the clinical setting
may enable a patient-oriented precision strategy for treatment of
childhood AML.
Supplementary Material
HaloPlex custom panels used in the
present study.
Quality metrics of next-generation
sequencing.
Candidate germline variants detected
in the present study.
Acknowledgements
Not applicable.
Funding
This present study was supported by grants from
MEXT/JSPS KAKENHI (grant no. JP 20K08157).
Availability of data and materials
The datasets generated and/or analyzed during the
present study could not be submitted to a public curated database
as the informed consent obtained does not include unrestricted
disclosure of sequencing data. Instead, these data are available
from the corresponding author upon reasonable request.
Authors' contributions
HI and AS wrote the manuscript. HI, MA, TM, MS and
AS performed the genetic analysis and interpreted the results. HI,
AI, RN, DK and AS performed patient care and collected the clinical
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Institutional
Review Board of Okayama University Hospital (Okayama, Japan).
Informed consent was obtained from the patients and/or their legal
guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Taga T, Tomizawa D, Takahashi H and Adachi
S: Acute myeloid leukemia in children: Current status and future
directions. Pediatr Int. 58:71–80. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rasche M, Zimmermann M, Borschel L,
Bourquin JP, Dworzak M, Klingebiel T, Lehrnbecher T, Creutzig U,
Klusmann JH and Reinhardt D: Successes and challenges in the
treatment of pediatric acute myeloid leukemia: A retrospective
analysis of the AML-BFM trials from 1987 to 2012. Leukemia.
32:2167–2177. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pikman Y and Stegmaier K: Targeted therapy
for fusion-driven high-risk acute leukemia. Blood. 132:1241–1247.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dohner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zwaan CM, Kolb EA, Reinhardt D,
Abrahamsson J, Adachi S, Aplenc R, De Bont ES, De Moerloose B,
Dworzak M, Gibson BE, et al: Collaborative efforts driving progress
in pediatric acute myeloid leukemia. J Clin Oncol. 33:2949–2962.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kolb EA and Meshinchi S: Acute myeloid
leukemia in children and adolescents: Identification of new
molecular targets brings promise of new therapies. Hematology.
2015:507–513. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kayser S and Levis MJ: Advances in
targeted therapy for acute myeloid leukaemia. Br J Haematol.
180:484–500. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ding L, Ley TJ, Larson DE, Miller CA,
Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan
MD, et al: Clonal evolution in relapsed acute myeloid leukaemia
revealed by whole-genome sequencing. Nature. 481:506–510.
2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wong TN, Ramsingh G, Young AL, Miller CA,
Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, et
al: Role of TP53 mutations in the origin and evolution of
therapy-related acute myeloid leukaemia. Nature. 518:552–555.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Quek L, Ferguson P, Metzner M, Ahmed I,
Kennedy A, Garnett C, Jeffries S, Walter C, Piechocki K, Timbs A,
et al: Mutational analysis of disease relapse in patients
allografted for acute myeloid leukemia. Blood Adv. 1:193–204.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Papaemmanuil E, Gerstung M, Bullinger L,
Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F,
Bolli N, et al: Genomic classification and prognosis in acute
myeloid leukemia. N Engl J Med. 374:2209–2221. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Greif PA, Hartmann L, Vosberg S, Stief SM,
Mattes R, Hellmann I, Metzeler KH, Herold T, Bamopoulos SA, Kerbs
P, et al: Evolution of cytogenetically normal acute myeloid
leukemia during therapy and relapse: An exome sequencing study of
50 patients. Clin Cancer Res. 24:1716–1726. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shiba N, Yoshida K, Shiraishi Y, Okuno Y,
Yamato G, Hara Y, Nagata Y, Chiba K, Tanaka H, Terui K, et al:
Whole-exome sequencing reveals the spectrum of gene mutations and
the clonal evolution patterns in paediatric acute myeloid
leukaemia. Br J Haematol. 175:476–489. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bolouri H, Farrar JE, Triche T Jr, Ries
RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, et al:
The molecular landscape of pediatric acute myeloid leukemia reveals
recurrent structural alterations and age-specific mutational
interactions. Nat Med. 24:103–112. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Tsukimoto I, Tawa A, Horibe K, Tabuchi K,
Kigasawa H, Tsuchida M, Yabe H, Nakayama H, Kudo K, Kobayashi R, et
al: Risk-stratified therapy and the intensive use of cytarabine
improves the outcome in childhood acute myeloid leukemia: The AML99
trial from the Japanese childhood AML cooperative study group. J
Clin Oncol. 27:4007–4013. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tomizawa D, Tawa A, Watanabe T, Saito AM,
Kudo K, Taga T, Iwamoto S, Shimada A, Terui K, Moritake H, et al:
Excess treatment reduction including anthracyclines results in
higher incidence of relapse in core binding factor acute myeloid
leukemia in children. Leukemia. 27:2413–2416. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hanada T, Kanamitsu K, Chayama K, Miyamura
T, Kanazawa Y, Muraoka M, Washio K, Imada M, Kageyama M, Takeuchi
A, et al: A Long-term survivor after congenital acute myeloid
leukemia with t(8 ; 16)(p11 ; p13). Acta Med Okayama. 70:31–35.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Iwasaki Y, Nishiuchi R, Aoe M, Takahashi
T, Watanabe H, Tokorotani C, Kikkawa K and himada A: Positive
minimal residual disease of FLT3-ITD before hematopoietic stem cell
transplantation resulted in a poor prognosis of an acute myeloid
leukemia. Acta Med Okayama. 71:79–83. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Green RC, Berg JS, Grody WW, Kalia SS,
Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond
KE, et al: ACMG recommendations for reporting of incidental
findings in clinical exome and genome sequencing. Genet Med.
15:565–574. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–423. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kalia SS, Adelman K, Bale SJ, Chung WK,
Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al:
Recommendations for reporting of secondary findings in clinical
exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy
statement of the american college of medical genetics and genomics.
Genet Med. 19:249–255. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ishida H, Kanamitsu K, Washio K, Muraoka
M, Sakakibara K, Matsubara T, Kanzaki H and Shimada A: Relapsed
infant MLL-rearranged acute lymphoblastic leukemia with additional
genetic alterations. Pediatr Blood Cancer. 63:2059–2060.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aoe M, Ishida H, Matsubara T, Karakawa S,
Kawaguchi H, Fujiwara K, Kanamitsu K, Washio K, Okada K, Shibakura
M and Shimada A: Simultaneous detection of ABL1 mutation and IKZF1
deletion in Philadelphia chromosome-positive acute lymphoblastic
leukemia using a customized target enrichment system panel. Int J
Lab Hematol. 40:427–436. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ishida H, Iguchi A, Aoe M, Takahashi T,
Tamefusa K, Kanamitsu K, Fujiwara K, Washio K, Matsubara T,
Tsukahara H, et al: Panel-based next-generation sequencing
identifies prognostic and actionable genes in childhood acute
lymphoblastic leukemia and is suitable for clinical sequencing. Ann
Hematol. 98:657–668. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sunami K, Ichikawa H, Kubo T, Kato M,
Fujiwara Y, Shimomura A, Koyama T, Kakishima H, Kitami M,
Matsushita H, et al: Feasibility and utility of a panel testing for
114 cancer-associated genes in a clinical setting: A hospital-based
study. Cancer Sci. 110:1480–1490. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nakayama H, Tabuchi K, Tawa A, Tsukimoto
I, Tsuchida M, Morimoto A, Yabe H, Horibe K, Hanada R, Imaizumi M,
et al: Outcome of children with relapsed acute myeloid leukemia
following initial therapy under the AML99 protocol. Int J Hematol.
100:171–179. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Duployez N, Marceau-Renaut A, Boissel N,
Petit A, Bucci M, Geffroy S, Lapillonne H, Renneville A, Ragu C,
Figeac M, et al: Comprehensive mutational profiling of core binding
factor acute myeloid leukemia. Blood. 127:2451–2459.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Faber ZJ, Chen X, Gedman AL, Boggs K,
Cheng J, Ma J, Radtke I, Chao JR, Walsh MP, Song G, et al: The
genomic landscape of core-binding factor acute myeloid leukemias.
Nat Genet. 48:1551–1556. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
de Rooij JDE, Branstetter C, Ma J, Li Y,
Walsh MP, Cheng J, Obulkasim A, Dang J, Easton J, Verboon LJ, et
al: Pediatric non-Down syndrome acute megakaryoblastic leukemia is
characterized by distinct genomic subsets with varying outcomes.
Nat Genet. 49:451–456. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Alexander TB, Gu Z, Iacobucci I, Dickerson
K, Choi JK, Xu B, Payne-Turner D, Yoshihara H, Loh ML, Horan J, et
al: The genetic basis and cell of origin of mixed phenotype acute
leukaemia. Nature. 562:373–379. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Döhner H, Estey E, Grimwade D, Amadori S,
Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA,
et al: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood.
129:424–447. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Marceau-Renaut A, Duployez N, Ducourneau
B, Labopin M, Petit A, Rousseau A, Geffroy S, Bucci M, Cuccuini W,
Fenneteau O, et al: Molecular profiling defines distinct prognostic
subgroups in childhood AML. Hemasphere. 2(e31)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shiba N, Yoshida K, Hara Y, Yamato G,
Shiraishi Y, Matsuo H, Okuno Y, Chiba K, Tanaka H, Kaburagi T, et
al: Transcriptome analysis offers a comprehensive illustration of
the genetic background of pediatric acute myeloid leukemia. Blood
Adv. 3:3157–3169. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Meshinchi S, Alonzo TA, Stirewalt DL,
Zwaan M, Zimmerman M, Reinhardt D, Kaspers GJ, Heerema NA, Gerbing
R, Lange BJ and Radich JP: Clinical implications of FLT3 mutations
in pediatric AML. Blood. 108:3654–3661. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Shimada A, Taki T, Tabuchi K, Taketani T,
Hanada R, Tawa A, Tsuchida M, Horibe K, Tsukimoto I and Hayashi Y:
Tandem duplications of MLL and FLT3 are correlated with poor
prognoses in pediatric acute myeloid leukemia: A study of the
Japanese childhood AML cooperative study group. Pediatr Blood
Cancer. 50:264–269. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ivey A, Hills RK, Simpson MA, Jovanovic
JV, Gilkes A, Grech A, Patel Y, Bhudia N, Farah H, Mason J, et al:
Assessment of minimal residual disease in Standard-risk AML. N Engl
J Med. 374:422–433. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Morita K, Kantarjian HM, Wang F, Yan Y,
Bueso-Ramos C, Sasaki K, Issa GC, Wang S, Jorgensen J, Song X, et
al: Clearance of somatic mutations at remission and the risk of
relapse in acute myeloid leukemia. J Clin Oncol. 36:1788–1797.
2018.PubMed/NCBI View Article : Google Scholar
|