Introduction
Nail-Patella syndrome (NPS) or hereditary
onycho-osteodysplasia (OMIM entry no. 161200) is a rare autosomal
dominant disease that affects multiple organs including the limbs,
skeleton, kidneys and eyes (1). The
classical clinical manifestations consist of nail dysplasia,
hypoplasia or absence of the patella, elbow dysplasia and iliac
horns (1). Renal involvement occurs
in 30-50% of patients with NPS and is the primary determinant of
prognosis (2,3). Renal manifestations typically include
proteinuria with or without hematuria, and nephrotic syndrome
(5-10% of cases), which progresses to end-stage renal disease
(ESRD) over varying periods of time (3). Additional relevant characteristics
observed in patients with NPS include open angle glaucoma and
ocular hypertension, which usually develop during adulthood and
occur in ~10 and 7% of patients, respectively (2,4). The
gene associated with NPS, LMX1B, maps on to chromosome
9q33.3 and contains 8 exons that encode a LIM-homeodomain
transcription factor called LMXB1 (5-7).
This protein belongs to the LIM-homeodomain protein family, and
serves an essential role in the normal development of
dorsal-ventral limb structures, morphogenesis, and in the functions
of podocytes and the glomerular basal membrane, anterior segment of
the eye, and some types of neurons (1,5).
LMX1B contains two cysteine-rich zinc-binding motifs
(LIM-A and LIM-B domains) at the N-terminus, which are all involved
in protein-protein interactions; a homeodomain in the middle
responsible for the interaction with specific DNA sequences in
target genes, and a C-terminal glutamine-rich domain of unknown
function (1,7). Most loss-of-function mutations of LMX1B
are clustered in the LIM domains and the homeodomain, encoded by
exons 2-3 and 4-6, respectively (8-11).
Several studies have reported entire deletions of the LMX1B
gene in patients with NPS, supporting haploinsufficiency as the
major pathogenic mechanism underlying the disease (8,9,12,13).
However, a clear genotype-phenotype correlation among NPS patients
is still lacking (8). Even patients
within the same family present a varying range of symptoms and
symptom severity, emphasizing the potential contribution of
modifier genes in the manifestation of the phenotype (11).
In the present case report, an unusual case of a
2-year old girl from Syria who presented with nephrotic syndrome
that evolved rapidly to ESRD is described. She exhibited classical
NPS symptoms including dystrophic nails and absence of patellae.
Using DNA sequencing analysis, a novel heterozygous missense
mutation in LMX1B (c.709T>C, p.S237P), which affected the
homeodomain of the LMX1B protein was identified, which was
predicted to be pathogenic.
Case report
Subject
The patient was a 2-year-old girl from Syria. She
was admitted to the Hospital General Univesitario Gregorio Marañón
(Madrid, Spain) for conventional hemodialysis. Genetic studies were
performed after obtaining written consent from the parents. The
Ethics Committee of Hospital Universitario Nuestra Señora de
Candelaria (Santa Cruz de Tenerife, Spain) approved the present
study. The clinical study included radiological examination, biopsy
and an ophthalmological exam.
Clinical manifestations
The patient presented with nephrotic syndrome at the
age of 2 years, which did not respond to steroid treatment. The
family history was unremarkable and the patient has one healthy
sister 2 years older than her. She was born after a normal
pregnancy; her weight and height were 3.8 kg and 52 cm,
respectively. She received breastfeeding and showed adequate
psychomotor development. A renal biopsy performed when she was 2
years and 2 months old revealed membranoproliferative
glomerulonephritis type I. She was diagnosed with steroid-resistant
nephrotic syndrome in the context of NPS. Therefore, corticosteroid
treatment was discontinued gradually. After the gradual progression
of the disease a second biopsy showed evolution to
glomerulosclerosis and glomerular fibrosis. The patient progressed
rapidly to ESRD 8 months after the first signs of renal impairment.
She was placed under conventional hemodialysis and 2 months later
under peritoneal dialysis until she received a renal transplant
when she was 4 years old. Recently, the patient reentered
hemodialysis due to chronic graft rejection and was transplanted
again. The orthopedic signs of NPS exhibited by the patient were
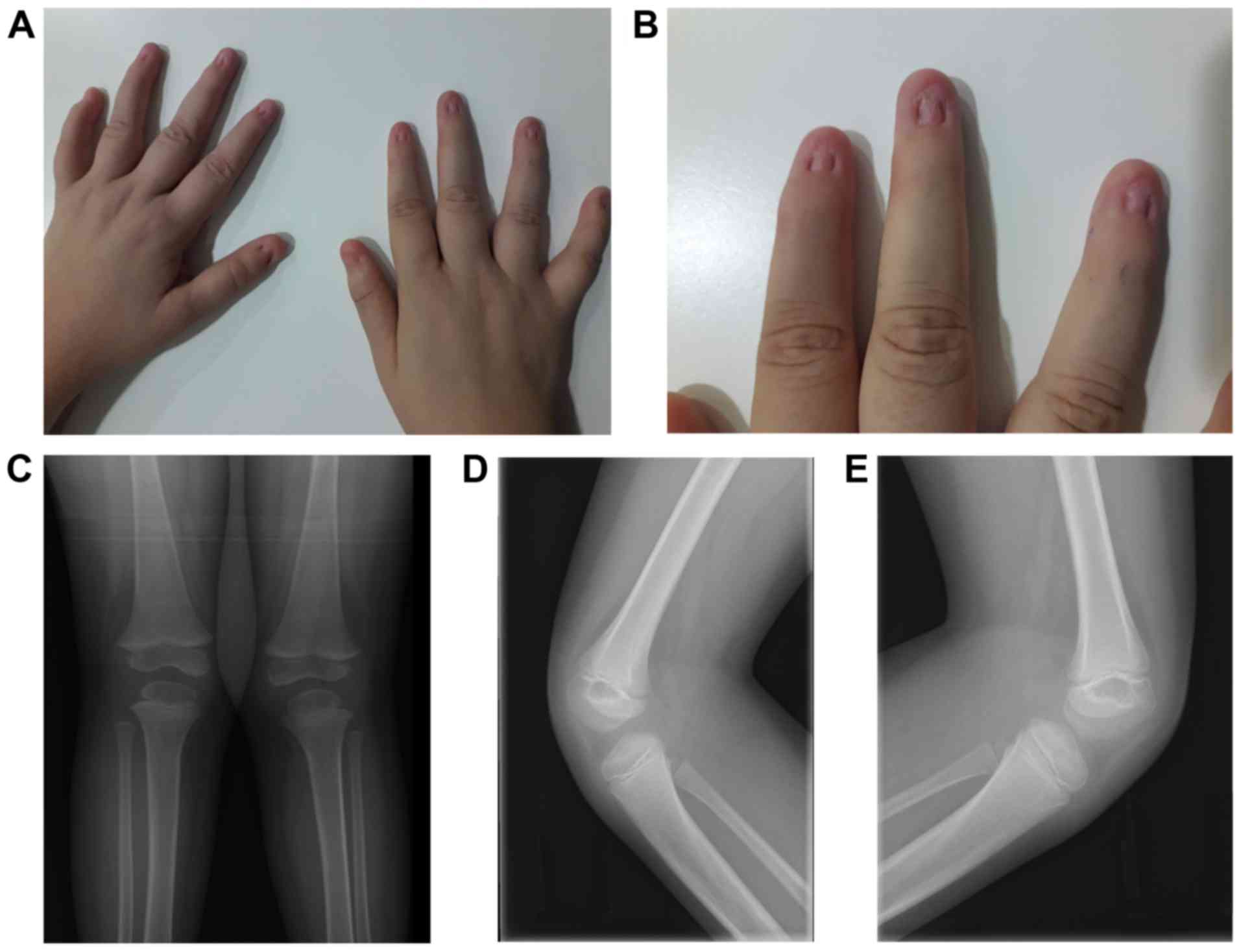
dystrophic nails and absence of patellae observed by X-ray
examination (Fig. 1). The patient
was noted to have coxa valga on X-ray (data not shown), although
this is not a characteristic of NPS. Ophthalmological examination
showed no ocular defects.
Materials and methods
DNA amplification and Sanger
sequencing
Genomic DNA was extracted from peripheral blood
samples using the GenElute Blood Genomic DNA kit according to the
manufacturer's protocol (Sigma-Aldrich; Merck KGaA). DNA quality
and quantity were assessed using a NanoDrop Lite spectrophotometer
(Thermo Fisher Scientific, Inc.). For analysis of mutations in the
LMX1B gene, the 8 coding exons and their intronic flanking
sequences were amplified by PCR as previously described (14-15). PCR products
were analyzed by agarose gel (1.5%) electrophoresis, and the
fragments were purified using QIAquick PCR purification kit
(Qiagen, Inc.). DNA sequencing of purified products was performed
by Macrogen Inc. Variants were identified by comparison to the
respective LMX1B reference sequences (GeneBank accession no.
NG_17039.1), and confirmed by sequencing additional independent
amplification products.
Bioinformatics analysis
Several databases, including gnomAD (gnomad.broadinstitute.org/), 1000 Genomes Project
(1000genomes.org/), Human Gene Mutation Database (HGMD,
hgmd.cf.ac.uk/ac/index.php) and ClinVar
(ncbi.nlm.nih.gov/clinvar/), were
explored to determine whether the mutation detected in the present
study had been reported previously. Online bioinformatics tools
PolyPhen-2 (genetics.bwh.harvard.edu/pph2), Align GVGD (agvgd.iarc.fr), MutPred2 (mutpred.mutdb.org), Mutation Taster (mutationtaster.org) and VarSome (varsome.com), a new search engine for human genomic
variation (16), were used to
predict the pathogenicity of the identified mutation. The protein
sequence of human LMX1B (isoform 2 containing 402 amino acids) was
obtained from the NCBI database (accession no. NP_001167618.1). The
protein structure homology-modelling server SWISS-MODEL was used to
model the 3D structure of the wild-type and mutant LMX1B
homeodomains using LIM/Homeobox protein Lhx4 as a template.
Results
Based on the orthopedic manifestations and the renal
course of the patient, mutational analysis of the LMX1B gene
was performed. The results of the DNA sequencing analysis showed a
novel heterozygous variant in exon 4 of LMX1B, c.709T>C
(g.78849T>C, genomic location Chr9; 129455570>C GRCh37;
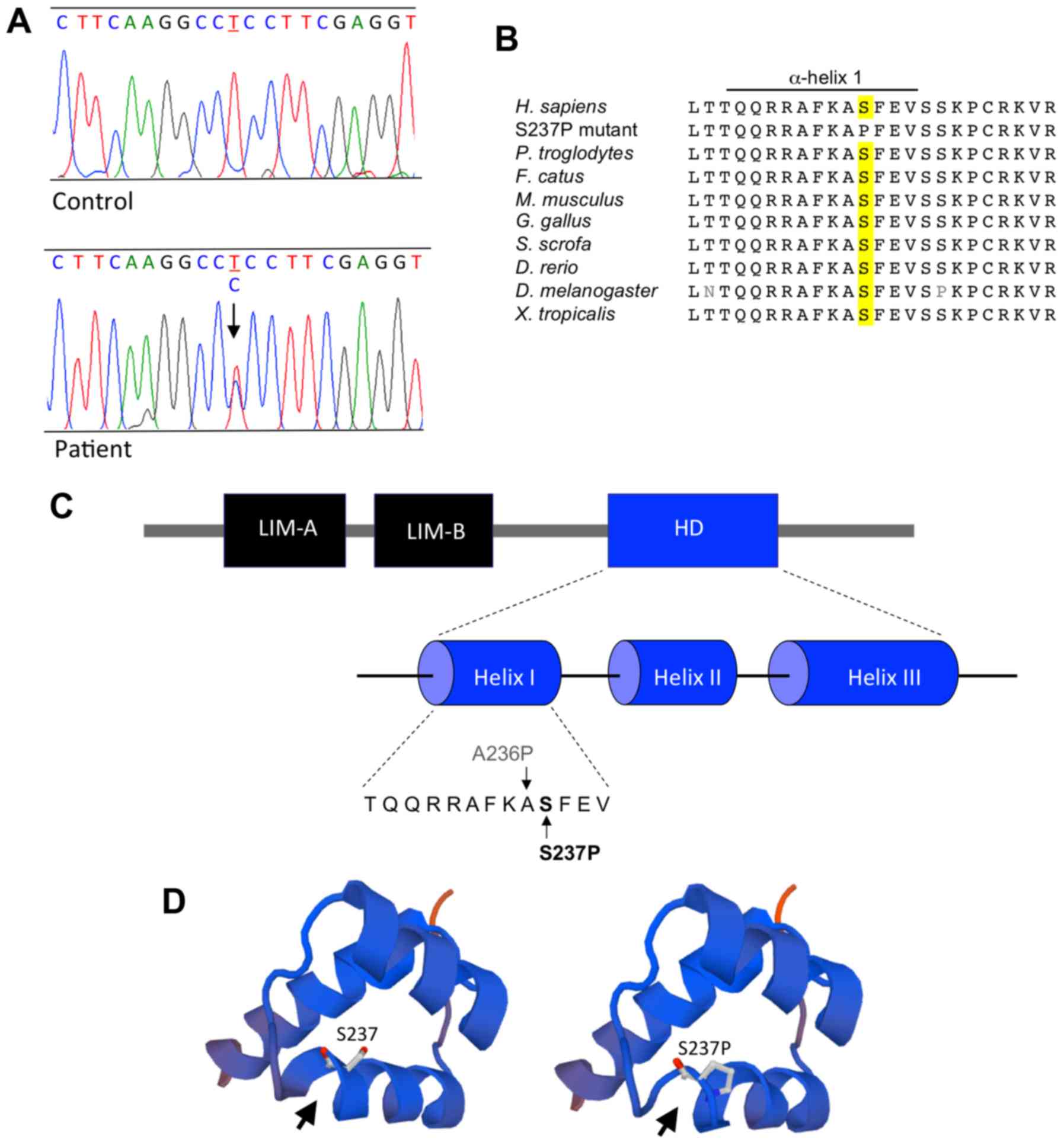
Fig. 2A). This nucleotide
substitution was predicted change a serine residue to proline at
position 237 (p.S237P) in the homeodomain of the LMX1B protein
(Ensembl transcript ID, ENST00000355497). Since blood samples from
the patient's parents were not available, it was not possible to
determine if the mutation was inherited from one of parents or if
it originated de novo. Variant c.709T>C, p.S237P was
absent from several databases, including gnomAD, 1000 Genomes
Project, HGMD and ClinVar. Homology analysis showed that serine 237
of LMX1B is conserved amongst different species (Fig. 2B). Analysis of p.S237P with five
different bioinformatics tools predicted that this variant was
pathogenic (Table I). VarSome
classified this substitution as likely pathogenic as it met 2
moderate (PM2 and PM3) and 3 supporting (PP2, PP3 and PP5)
pathogenicity criteria established by the American College of
Medical Genetics and Genomics (17).
The novel LMX1B mutation described here was submitted to
ClinVar (accession no. VCV000694525; variant, NM_002316.4;
ncbi.nlm.nih.gov/clinvar/variation/694525/). As shown
in Fig. 2C, the missense mutation
p.S237P is located in α-helix 1 of the LMX1B homeodomain. To
evaluate the structural impact of this mutation, 3D models of the
wild-type and mutant homeodomains were constructed using
SWISS-MODEL. The results suggested that the substitution of serine
for proline at position 237 transformed the segment A236-S237-F238
of the α-helix 1 in the homeodomain into a strand (Fig. 2D).
 | Table IPathogenicity prediction of the
p.S237P mutation in LMX1B using various bioinformatics tools. |
Table I
Pathogenicity prediction of the
p.S237P mutation in LMX1B using various bioinformatics tools.
| Outcomes |
PolyPhen-2a | Align
GVGDb | MutPred2c | Mutation
Tasterd | VarSomee |
|---|
| Score | 1.0 | Class 65 | 0.886 | 74 | - |
| Prediction | Probably
damaging | Most likely
damaging | Likely
pathogenic | Disease causing | Likely
pathogenic |
Discussion
NPS is an autosomal dominant disorder characterized
by dysplastic nails, a hypoplastic or absent patella, and elbow
dysplasia (1,2). In some cases, patients may also exhibit
differing levels of proteinuria that can progress to ESRD, and less
frequently open-angle glaucoma. The skeletal disorders can be
detected at birth, but in some cases they are less significant,
making the diagnosis problematic (1,3). In the
present case report, NPS was challenging to diagnose immediately as
nephrotic syndrome developed before obvious orthopedic symptoms,
and there was no family history of NPS. This case was quite
uncommon as the patient presented persistent nephrotic syndrome
very early, at the age of 2 years, and developed rapidly to ESRD.
The majority of patients with NPS exhibit a slow gradual loss of
renal function, and only a small number of cases, where a rapid
progression to ESRD at a young age, have been reported (15,18). The
factors responsible for progression to ESRD in NPS are unknown, but
it is possible that potential modifying genes may contribute to it.
The nephropathy in the patient reported on in the present case
report was treated with corticosteroids and diuretics, and the
corticoresistance supported the association of the nephrotic
syndrome with NPS. Moreover, renal biopsy of the patient revealed a
characteristic irregular thickening of the glomerular basement
membrane with progression to fibrosis and glomerulosclerosis
observed in other patients with NPS (3). Additionally, the nail and skeletal
phenotype of the patient was consistent with previously published
data of NPS (2).
NPS is caused by loss-of-function mutations in the
LMX1B gene, which is expressed in several different tissues
implicated in the clinical phenotype (5-7). In
the kidney, LMX1B is expressed in podocytes and regulates the
expression of several genes required for correct podocyte
differentiation and function (1).
Studies using inducible podocyte specific Lmx1b knockout
mice have established the importance of LMX1B in fully
differentiated podocytes, and suggest that this transcription
factor is essential for the maintenance of a properly structured
actin cytoskeleton (19).
Additionally, LMX1B has been shown to bind enhancer sequences (FLAT
elements) in the promoter regions of ABRA and ARL4C,
two genes encoding actin cytoskeleton-associated proteins (19). These studies concluded that podocyte
pathogenesis in NPS likely results from a dysregulation of the
actin cytoskeleton.
During embryogenesis, LMX1B expression is high in
the dorsal mesenchyme of developing limbs and is involved in
dorso-ventral patterning (20).
Following the formation of the dorsal-ventral axis, LMX1B activates
the expression of several dorsalizing genes, represses the
expression of ventralizing genes, and regulates the development of
dorsal distal limb structures, such as the nails and patellas
(20). Therefore, the skeletal
phenotype of patients with NPS is the result of a defect in
dorsoventral patterning caused by LMX1B mutations.
LMX1B is also expressed during ocular development in
the anterior portions of the murine eye (21); ~10% of NPS patients develop
open-angle glaucoma and an additional 7% exhibit ocular
hypertension (2,8). These ocular symptoms usually develop
during adulthood. The patient reported in the present case report
is now 14 years old and she has not yet developed glaucoma. She
undergoes ophthalmological reviews every 6 months. The pathogenesis
of glaucoma in NPS is still unknown, but homozygous lmx1b
knockout mice display iris and ciliary body hypoplasia and corneal
collagen fibril defects, suggesting a role for LMX1B in collagen
regulation during development (21).
Several of the mutations identified in patients with
NPS are localized in the homeodomain of LMX1B (8,9,11). This highly conserved DNA-binding
motif forms part of several transcription factors present in
numerous organisms, from yeast to humans (22). These transcription factors serve
essential roles during development and adult homeostasis, and
mutations in their genes are known to cause human diseases. These
genes include HOXA13 and HOXD13, which are associated
with skeletal defects in distal extremities; LHX3 and
POU1F1, which are associated with pituitary hormone
deficiency; and PAX6 which is associated with eye-related disorders
(22). The homeodomain is comprised
of ~60 amino acids and includes three α-helices, of which helices 2
and 3 adopt a helix-turn-helix motif, and a flexible N-terminal
arm. Helix 3 and the N-terminal arm interact directly with the
major groove and the adjacent minor groove of target DNAs,
respectively, whereas helices 1 and 2 maintain the tertiary
structure important in the helix-turn-helix configuration.
Mutational analysis of LMX1B in the present case report
identified a novel heterozygous variant, c.709T>C, p.S237P, that
explained the phenotype observed. This variant, which was not found
in any of the reference databases, resulted in a change in a
conserved serine residue to a helix-disrupting proline residue, and
was predicted to be pathogenic by several different bioinformatics
tools. The evolutionary conservation of serine 237 indicates that
this residue is essential for the structure and function of the
LMX1B homeodomain. Although mutational analysis of the patient's
parents was not possible, it is hypothesized that p.S237P is a
de novo mutation as there was no history of nephropathy,
skeletal defects or nail dystrophy in either parent, sister,
grandparents or uncles. The identified mutation is located next to
the amino acid residue affected by recurrent mutation c.706G>C,
p.A236P in α-helix 1 of the homeodomain and appears as p.A213P in
(6). p.A236P has been shown to
abolish both DNA binding of the LMX1B homeodomain to the FLAT
element of the insulin promoter and transactivation of a reporter
gene (6,12). Knoers et al (23) indicated that p.A236P disrupted the 3D
structure of the homeodomain and, therefore, disrupted DNA binding.
Similarly, p.S237P also introduces a proline residue that may
destabilize the helix conformation and affect the overall tertiary
structure of the homeodomain. Analysis of the predicted structural
model of the mutant homeodomain in the present study indicated that
p.S237P indeed disturbed the carboxy-terminal segment of α-helix 1.
Thus, together, these results suggests that p.S237P destabilizes
α-helix 1, abolishing DNA binding and the subsequent
transcriptional activity of LMX1B. This hypothesis requires
confirmation using functional studies.
In conclusion, a novel missense mutation
(c.709T>C, p.S237P) in the LMX1B gene of a child
presenting with nephrotic syndrome and rapid progression to ESRD
was identified. Based on in silico structural analysis of
this mutation, the α-helix 1 was altered, which abrogated binding
of the LMX1B homeodomain to the target DNA. This report further
expands on the mutational spectrum of LMX1B, and provides
insight into the molecular mechanisms of NPS pathology.
Acknowledgements
Not applicable.
Funding
This work was supported by integrated in the Plan
Nacional de I+D+I 2013-2016 (grant no. PI17/00153), and co-financed
by the ISCIII-Subdirección General de Evaluación y Fomento de la
Investigación and the European Regional Development Fund ‘Another
way to build Europe’.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. The novel LMX1B mutation described in the present
has been submitted to ClinVar (accession no. VCV000694525; variant,
NM_002316.4.
Authors' contributions
SC and APR performed PCR amplifications and analyzed
the DNA sequences. SC, APR and FCM carried out the bioinformatics
analysis. OAB collected the patient's data and followed up the
case. SC and FCM wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Hospital Universitario Nuestra Señora de Candelaria (Santa Cruz de
Tenerife, Spain) (approval no. C.P. MO PI17/00153-C.I. PI56-17).
Written informed consent for participation of the child in the
genetic study was obtained from her parents.
Patient consent for publication
Written informed consent for publication of this
study was obtained from the parents of the child.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Witzgall R: Nail-patella syndrome.
Pflugers Arch. 469:927–936. 2017.
|
|
2
|
McIntosh I, Dunston JA, Liu L, Hoover-Fong
JE and Sweeney E: Nail patella syndrome revisited: 50 years after
linkage. Ann Hum Genet. 69:349–363. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lemley KV: Kidney disease in nail-patella
syndrome. Pediatr Nephrol. 24:2345–2354. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lichter PR, Richards JE, Downs CA,
Stringham HM, Boehnke M and Farley FA: Cosegregation of open-angle
glaucoma and the nail-patella syndrome. Am J Ophthalmol.
124:506–515. 1997.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dreyer SD, Zhou G, Baldini A, Winterpacht
A, Zabel B, Cole W, Johnson RL and Lee B: Mutations in LMX1B cause
abnormal skeletal patterning and renal dysplasia in nail patella
syndrome. Nat Genet. 19:47–50. 1998.PubMed/NCBI View Article : Google Scholar
|
|
6
|
McIntosh I, Dreyer SD, Clough MV, Dunston
JA, Eyaid W, Roig CM, Montgomery T, Ala-Mello S, Kaitila I,
Winterpacht A, et al: Mutation analysis of LMX1B gene in
nail-patella syndrome patients. Am J Hum Genet. 63:1651–1658.
1998.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Vollrath D, Jaramillo-Babb VL, Clough MV,
McIntosh I, Scott KM, Lichter PR and Richards JE: Loss-of-function
mutations in the LIM-homeodomain gene, LMX1B, in nail-patella
syndrome. Hum Mol Genet. 7:1091–1098. 1998.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bongers EM, Huysmans FT, Levtchenko E, de
Rooy JW, Blickman JG, Admiraal RJ, Huygen PL, Cruysberg JR, Toolens
PA, Prins JB, et al: Genotype-phenotype studies in nail-patella
syndrome show that LMX1B mutation location is involved in the risk
of developing nephropathy. Eur J Hum Genet. 13:935–946.
2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Marini M, Bocciardi R, Gimelli S, Di Duca
M, Divizia MT, Baban A, Gaspar H, Mammi I, Garavelli L, Cerone R,
et al: A spectrum of LMX1B mutations in Nail-Patella syndrome: New
point mutations, deletion, and evidence of mosaicism in unaffected
parents. Genet Med. 12:431–439. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hamlington JD, Jones C and McIntosh I:
Twenty-two novel LMX1B mutations identified in nail patella
syndrome (NPS) patients. Hum Mutat. 18(458)2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ghoumid J, Petit F, Holder-Espinasse M,
Jourdain AS, Guerra J, Dieux-Coeslier A, Figeac M, Porchet N,
Manouvrier-Hanu S and Escande F: Nail-Patella Syndrome: Clinical
and molecular data in 55 families raising the hypothesis of a
genetic heterogeneity. Eur J Hum Genet. 24:44–50. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dreyer SD, Morello R, German MS, Zabel B,
Winterpacht A, Lunstrum GP, Horton WA, Oberg KC and Lee B: LMX1B
transactivation and expression in nail-patella syndrome. Hum Mol
Genet. 9:1067–1074. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sato U, Kitanaka S, Sekine T, Takahashi S,
Ashida A and Igarashi T: Functional characterization of LMX1B
mutations associated with nail-patella syndrome. Pediatr Res.
57:783–788. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Claverie-Martin F, Trindade A,
Garcia-Gonzalez NC and Callejon AC: Novel missense mutation
affecting the LIM-A domain of LMX1B in a family with Nail-Patella
syndrome. Intractable Rare Dis Res. 8:14–19. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lee BH, Cho TJ, Choi HJ, Kang HK, Lim IS,
Park YH, Ha IS, Choi Y and Cheong HI: Clinico-genetic study of
nail-patella syndrome. J Korean Med Sci. 24 (Suppl 1):S82–S86.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kopanos C, Tsiolkas V, Kouris A, Chapple
CE, Albarca Aguilera M, Meyer R and Massouras A: VarSome: The human
genomic variant search engine. Bioinformatics. 35:1978–1980.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bongers EM, Gubler MC and Knoers NV:
Nail-patella syndrome. Overview on clinical and molecular findings.
Pediatr Nephrol. 17:703–712. 2002.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Burghardt T, Kastner J, Suleiman H,
Rivera-Milla E, Stepanova N, Lottaz C, Kubitza M, Böger CA, Schmidt
S, Gorski M, et al: LMX1B is essential for the maintenance of
differentiated podocytes in adult kidneys. J Am Soc Nephrol.
24:1830–1848. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen H, Lun Y, Ovchinnikov D, Kokubo H,
Oberg KC, Pepicelli CV, Gan L, Lee B and Johnson RL: Limb and
kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B
in human nail patella syndrome. Nat Genet. 19:51–55.
1998.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pressman CL, Chen H and Johnson RL: LMX1B,
a LIM homeodomain class transcription factor, is necessary for
normal development of multiple tissues in the anterior segment of
the murine eye. Genesis. 26:15–25. 2000.PubMed/NCBI
|
|
22
|
Chi YI: Homeodomain revisited: A lesson
from disease-causing mutations. Hum Genet. 116:433–444.
2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Knoers NV, Bongers EM, van Beersum SE,
Lommen EJ, van Bokhoven H and Hol FA: Nail-patella syndrome:
Identification of mutations in the LMX1B gene in Dutch families. J
Am Soc Nephrol. 11:1762–1766. 2000.PubMed/NCBI
|