1. Rheumatoid arthritis and depression
Rheumatoid arthritis (RA) is a chronic autoimmune
disease that typically causes damage to the joints. The disease
primarily targets the synovial membrane, cartilage and bone. As a
systemic inflammatory disease, RA also affects other tissues and
organs, and is thus associated with progressive disability,
potential systemic complications, high socioeconomic costs, and
early death worldwide (1). Although,
there have been advances in the understanding of the pathogenesis
of RA, the cause of RA remains elusive (1). The pathogenesis of RA is complex and
involves large numbers of different cell types and signaling
pathways (2). It is widely accepted
that autoimmune processes and cytokines may serve important roles
in the initiation and perpetuation of the disease (3,4). Among
these, TNF, IL-6 and IL-1 are the best studied, and are further
discussed in the following section. The current strategies for
treatment of RA include symptomatic management and early disease
therapy (5). As RA is an
inflammatory disease, medications that suppress inflammation, such
as non-steroidal anti-inflammatory drugs and glucocorticoids are
frequently used as first-line therapeutic agents for treatment of
the symptoms (6). During the last
decade, disease-modifying antirheumatic drugs (DMARDs) were
introduced as an important therapeutic strategy due their effect on
the binding of pro-inflammatory cytokines to their corresponding
receptors (7), and these should be
initiated as soon as RA is diagnosed (8). The most commonly used biological DMARDs
in RA therapy include inhibitors of TNF-α (infliximab, etanercept,
adalimumab, golimumab and certolizumab pegol), IL-1 inhibitors,
anti-IL-6 receptor monoclonal antibody (tocilizumab), the T-cell
signaling inhibitor (abatacept) and the chimeric anti-CD20
monoclonal antibody (rituximab) (9).
Patients started on combined treatment of conventional synthetic
DMARDs with biological DMARDs in earlier stages showed obvious
clinical improvement and less damage to the joints in the BeSt
study (10).
Depression and anxiety are frequent comorbidities in
patients with RA and it has been shown that they may occur in up to
42% in RA patients (11). It is
estimated that ~30% of patients with RA develop depression within 5
years of being diagnosed (12).
Depression significantly and synergistically contributes to
increased mortality and morbidity in patients with RA, and also
increases health care costs for the health system (13). Inflammatory networks and cytokines
may constitute a link between depression and RA. The co-occurrence
of depression in RA is significantly associated with age, and
younger patients show a higher risk of developing depression
compared with older patients (12).
Low self-esteem is among the strongest predictors in patients with
RA who may eventually end up with depression (12). During the past two decades, the
reciprocal communication between the central nervous system (CNS),
endocrine system and immune system has been well established
(14). Some of the psychiatric
diseases, including depression, are hypothesized to be closely
associated with systemic inflammation and cytokines. Therefore,
exploring the common functions that may be shared by RA and
depression will both broaden the horizon of basic research in these
two fields and may also provide theoretical support for the
clinical treatment of RA with a systemic view.
The aim of this review is to discuss the biological
contribution and potential therapeutic targets of the major
cytokine families in the aetiology of RA and depression, and
summarize the complex pathophysiology of this disease with a focus
on cytokines, including TNF-α, IL-6 and IL-1 and their families and
relevant signaling pathways. Furthermore, the therapeutic agents
that are presently utilized in both of the diseases, along with
potential therapeutic targets are reviewed. Additionally, how an
improved understanding of the role of cytokines and signaling
pathways in these two diseases may help the development of novel
therapeutic strategies with specific targets is briefly
discussed.
2. Immunological pathology of RA
Major histocompatibility complex (MHC) is considered
a key factor associated with a predisposition to RA (15). Outside of the MHC region, additional
risk loci have been identified and validated including PTPN22,
STAT4, PADI4, CTLA4 and FcγRs, as well as various cytokine and
cytokine-receptor loci. Several immune modulators (cytokines and
effector cells) and signaling pathways involved in the
pathophysiology of RA have been revealed. The complex interactions
between the inflammatory cytokines are responsible for joint damage
that is initiated at the synovial membrane and then progresses to
other structures of the joint. There is extensive activation of
monocytes, macrophages, and synovial fibroblasts, as well as
overproduction of pro-inflammatory cytokines, such as IL-1, IL-6
and TNF-α amongst others (16).
There are other cytokines and chemokines that are also found in the
synovial membrane, such as IL-15, IL-17 and IL-18. These
inflammatory cytokines activate several signaling pathways and
induce transcription of the genes, which are key factors involved
in inflammation and tissue degradation (17). Due to the important roles of
inflammatory cytokines in RA, there has been increasing interest in
how the inflammatory cytokines within a complex regulatory network
are associated with disease progression of RA.
3. Inflammatory cytokines in RA
Inflammatory cytokines are responsible for
stimulating destructive mechanisms in the joint which results in
structural injury and subsequently leads to a functional decline in
mobility and disability.
Role of IL-1 and IL-18 in RA
IL-1 is an important inflammatory factor that is
involved in the pathogenesis of RA. IL-1 induces an inflammatory
response and activates or increases the expression of other
pro-inflammatory mediators that promote damage to the joints
(18). In the synovial membrane,
IL-1 and IL-1 receptor antagonists are abundantly expressed
(18). IL-1 activation causes
migration of inflammatory cells to the joints and the synovium in
patients with RA. Then, macrophages, lymphocytes, monocytes and
transformed fibroblasts are triggered to produce increased
quantities of IL-1, which contributes to the production and
secretion of various cellular messengers (19). Regulatory agents, such as
proteoglycans and proteases contribute to the formation and
accumulation of pannus in the joints, resulting in the erosion of
the cartilage and bone, fibrosis, and ultimately articular function
loss (20). At the cellular level,
there are several other cytokines induced by IL-1, including TNF-α
and IL-6(18). As the key
pro-inflammatory cytokines in RA, IL-1 and TNF-α act
synergistically in promoting further inflammation and matrix damage
to the arthritic joints (21). In an
experimental animal model, IL-1 was shown to stimulate cartilage
and bone re-absorption and inhibit articular collagen and
proteoglycan synthesis (22).
Experiments performed on RA animal models suggest that IL-1 serves
a crucial role in the degradation of proteoglycans and thus
increases joint damage (22,23). Local production of IL-1 was shown to
inhibit proteoglycan synthesis in the knee joints of a
collagen-induced arthritis (CIA) model (24). In addition, IL-6 in synovial cells
can lead to the acceleration of joint breakdown by attracting and
retaining large quantities of leukocytes in the inflamed synovium
(25). In patients with RA, there
are increased levels of IL-1 in the plasma and synovial fluid
(26,27). The IL-1 family includes IL-1α and
IL-1 and IL1 receptor antagonist (IL-1Ra). The biological
activation of cells by IL-1 requires the expression of the type I
IL-1 receptor (IL-1R). The receptor antagonist IL-1Ra, generated by
certain cells in the joint, can competitively bind with IL-1R. As
an anti-inflammatory protein that binds to IL-1R without
transducing signals, IL-1Ra may, to some extent, regulate the
activity of IL-1(18). It has been
reported that IL-1Ra-deficient mice develop spontaneous arthritis,
mediated through the amplification of Th17-dependent inflammation
(28). IL-1Ra effectively attenuated
joint destruction in an experimental RA model (29). There is a dysregulation of IL-1Ra
production and inhibition of the effects of IL-1β in RA patients
(30). IL-18, another member of the
IL-1 cytokine superfamily, is widely detected in the synovium of
patients with RA (18). The mRNA
expression levels of IL-18 and IL-1β are differentially regulated.
It has been found that IL-18 distributes in the synovial membrane
with a different pattern compared with IL-1β. While IL-1β is
localized primarily throughout the interstitium, lining layer and
vasculature, IL-18 is highly expressed in lymphocytic aggregates
(31). The expression of IL-18 in
synovial tissues in vitro is associated with increased TNF-α
and IL-1β, and the blockade of IL-18 by a neutralizing antibody was
shown to ameliorate the disease in rodent RA models (32). IL-18 also contributes to cartilage
degradation by affecting cell proliferation and the expression of
matrix metalloproteinase and inducible NO synthase gene expression
(33).
Role of IL-6 in RA
IL-6, which was initially identified as a B cell
regulatory factor, is now recognized as a soluble mediator with a
pleiotropic function on inflammation, immune response, and
hematopoiesis (34). It serves
important roles in RA pathogenesis via the activation and
maturation of B and T cells, as well as the production of
autoantibodies. IL-6 can stimulate B and T cell functions via
promoting the proliferation of plasmablastic precursors in the bone
marrow, maturating the immunoglobulin-producing plasma cells and
through its role in T cell activation and proliferation (35). Although certain cytokines work on
target cells that are close to their secretion site, IL-6 exerts
its effects on distant target cells through ubiquitously expressed
receptors. Classical IL-6 signaling requires a protein complex that
includes a membrane-bound, non-signaling α-receptor unit (IL-6R)
and two signal-transducing glycoprotein 130 (gp130) subunits
(36). Conversely, IL-6
trans-signaling involves binding between a soluble receptor
(sIL-6R) and membrane-bound gp130 subunits (36). As IL-6R expression is restricted to
only a few cell types, trans-signaling leads to an increase in the
range of IL-6-responsive cells (36). IL-6 trans-signaling is one of the
major factors involved in the pathogenesis of RA. It is
hypothesized that the increased IL-6 bioactivity is responsible for
the local and systemic effects of RA. It seems that IL-6 acts as a
major mediator of the acute phase response in RA (36). IL-6 is a 26-kDa pleiotropic cytokine,
encoded on chromosome 7, and is produced by various cell types,
including T cells and B cells (36).
It has been reported that IL-6 is one of the most abundantly
expressed inflammatory cytokines in the rheumatoid synovium
(37). The concentration of IL-6 and
sIL6R is correlated with the histological changes of chronic
synovitis in patients with RA, while localized increases in sIL-6R
are correlated with leukocyte infiltration (38). Vascular endothelial growth factor
(VEGF) is a potent angiogenic factor that promotes the migration
and proliferation of endothelial cells (39). VEGF also induces vascular
permeability and mediates inflammation. IL-6 was found to enhance
joint inflammation and damage in patients with RA by affecting VEGF
expression (40). IL-6 knockout mice
were protected from CIA and exhibited reduced immune cell responses
and tissue damage of knee joints (34,41).
IL-6 receptor blocking can ameliorate joint disease in murine CIA
(34).
Role of TNF-α in RA
TNF-α is a pleiotropic, pro-inflammatory cytokine
and is considered to serve a key role in the pathophysiological
processes of RA. TNF-α is a ligand of the TNF superfamily (42). TNF-α is inserted into the cell
membrane, and the extracellular portion is subsequently released
into circulation (43). There are
two distinct membrane receptors that have been identified and
cloned, namely TNF-R1 and TNF-R2. The apparent molecular weight
between the two receptors varies because of the variation in
glycosylation. These receptors can bind to both the soluble and
membrane-bound forms of TNF-α (44).
TNF-R1 is responsible for the majority of the cellular responses to
TNF-α (44). The ubiquitous
expression of TNF-R, along with cell-specific effector molecules
triggered by TNF-R contribute to the various effects of TNF-α
including apoptosis, the synthesis of protein and lipid
inflammatory molecules, and transcription factors (45). TNF-α contributes to activation of
other inflammatory cytokines, including IL-1, IL-6, IL-8, and
granulocyte-macrophage colony-stimulating factor (GM-CSF) (46). In an in vitro study, blocking
TNF-α decreased the levels of IL-1, IL-6, IL-8, and GM-CSF in
cultured synovial cells from RA patients (47). Thus, blockade of TNF-α may exert a
more comprehensive effect on inflammation than other cytokines,
such as IL-1, due to its high concentration in the synovial fluids.
TNF-α is suggested to serve a key role in rheumatoid synovitis. In
a CIA model, it was reported that TNF-α levels were elevated early
in the disease course of CIA and persisted at high levels
throughout the later stages (48).
Mice with downregulated TNF-α expression developed chronic
symmetric polyarthritis with histological features similar to that
in human RA (49,50).
4. Anti-cytokine therapies in RA
Recent developments in understanding the role of
cytokines in the pathological changes of RA, combined with
biopharmaceutical progress, have facilitated the development of
novel treatments.
Anti-TNF-α agents in RA
Studies have shown that blockade of TNF appears to
be the major breakthrough in RA treatment, and that TNF-α
antagonists significantly reduce the symptoms of RA in patients
(51,52). Clinical studies have shown that the
efficacy of the TNF inhibitors is at least comparable to
methotrexate, which is one of the most effective medications
currently used to treat RA (53).
Long-term anti-TNF-α treatment reversed endothelial dysfunction in
RA (54). A recent review indicated
that TNF-α antagonists can ameliorate the progression of
atherosclerosis and arterial changes (21,55). The
drugs currently available in the clinical practice and clinical
trials are biological protein-based drugs, such as antibodies to
TNF-α or agents targeting TNF-α receptors (for example, linked to
Fc dimers) (56). There are five
TNF-α inhibitors that have been approved for treatment of RA,
including infliximab, etanercept, adalimumab, certolizumab and
golimumab (57,58). Their practical use has been limited
by the need for repeated injections and other side effects
(59). A recent large US
observational study has also raised concerns regarding the risk of
skin cancer with the use of biological therapy (60).
Anti-IL-1 agent in RA
Blocking of IL-1 has been considered as a potential
strategy for treating RA. Anakinra, a recombinant form of
human IL-1R antagonist, appears to inhibit the proinflammatory
effect of IL-1(18). It is identical
to the natural endogenous IL-1RA form (61). Due to its short half-life, daily
injections are required. Anakinra decreases the migration of immune
cells into the joint of patients with RA (62). In RA patients, joint damage is
significantly attenuated following anakinra treatment (63). The most commonly observed adverse
effect of anakinra is the susceptibility to infections and skin
irritation at the injection site (64). However, due to the inconvenience of
daily injections of anakinra and the superior clinical effects of
other anti-cytokine therapies, anakinra is not used as a first-line
treatment in RA (63,64).
Anti-IL-6 agent in RA
A monoclonal antibody, Tocilizumab (TCZ) binds to
IL-6R and the binding subsequently blocks dimerization of the
receptor complex, preventing IL-6 transmembrane signaling (65). The efficacy of TCZ in the treatment
of RA has been confirmed in a few clinical trials (66,67).
Reducing IL-6 activity in the inflammatory process of RA prevents
inflammation of the joints, decreases damage, and reduces certain
systemic symptoms (68,69). The erosion in joint and space
narrowing were significantly decreased in TCZ treated patients
compared with the DMARD treated group, suggesting that the
inhibition of IL-6 was effective in both managing the symptoms and
sustaining structural integrity of the joints (70). Similar to other disease modifying
drugs, safety is always a major concern when investigating a new
line of therapies (71). Further
studies are required to examine the treatment benefits of TCZ
therapy in RA treatment (68).
Upcoming therapies for RA
There are several potentially promising therapies
targeting cytokines in RA which are being studied and may serve as
suitable novel therapies for RA treatment. IL-18 may be a novel
target for treating patients with RA. Recombinant IL-18 binding
protein treatment abrogated disease severity (29), and treatment of anti-IL-18 in mice
with collagen-induced arthritis exhibited similar results (29). IL-10 and IL-4 are cytokines with
opposing-regulatory roles, both of which could downregulate
pro-inflammatory responses. IL-10 and IL-4 are suggested to
suppress the generation of inflammatory cytokines including IL-1,
IL-6 and TNF-α in RA. A recent study on Janus kinase (JAK)
inhibitors demonstrated that the JAK/STAT signalling pathway
mediated cytokines important for disease by facilitating the
downstream signaling cascade of cytokine receptors upon binding
with their respective cytokines, such as by increasing
transcription of certain genes (72). For example, tofacitinib, a typical
JAK inhibitor that interferes with the JAK-STAT signaling pathway
and mRNA transcription, is currently used for the treatment of RA.
Previous studies have shown that stimulation with IFNs results in
STAT activation (73,74). Table I
summarizes the available anti-cytokine agents for treatment of
RA.
 | Table IAnti-cytokine based therapeutic
options for treatment of rheumatoid arthritis. |
Table I
Anti-cytokine based therapeutic
options for treatment of rheumatoid arthritis.
| Anti TNF-α
agents | Anti IL-1
agent | Anti IL-6
agent |
|---|
| Infliximab | Anakinra | Tocilizumab |
| Etanercept | | |
| Adalimumab | | |
| Certolizumab | | |
| Golimumab | | |
5. Role of cytokines in depression:
Mechanism and treatment
IL-6 in depression
It has previously been shown that IL-6 is associated
with stress-related disorders, including depression and anxiety
(75). A meta-analysis showed that
IL-6 is the most significantly increased cytokine in the serum of
depressed patients, and thus IL-6 levels in the serum may serve as
a promising predictive biomarker for diagnosis of depression
(75). Interestingly, a paediatric
study found that peripheral inflammation predicted the occurrence
of depression in younger patients (76). Children aged 9 with higher levels of
IL-6 demonstrated a 10% increased likelihood of suffering
depression at the age of 18 compared with children with lower
levels of IL-6. This result suggests that monitoring the blood IL-6
levels in younger patients at a high risk of depression during the
early stages may be used to implement measures which would prevent
the progression of deterioration of a patient's mood. Furthermore,
several monoclonal antibodies are currently in clinical trials for
their use in the possible treatment of mood disorders, such as
chimeric IL-6 receptor antibodies (Tocilizumab) or IL-6 antibodies
(Siltuximab/Sirukumab) (75).
IL-1 in depression
It is well-established that the hippocampus is one
of the key brain regions involved in the pathophysiology of
depression (77). Stress and other
environmental factors impact the functions of neurons in the
hippocampus and influences depressive and anxiety-like phenotypes
(77). Among the IL-1 cytokines,
IL-1β is considered a key mediator responding to stressors, and it
is widely expressed in the CNS, including the hypothalamus,
hippocampus, and cerebral cortex. A previous study showed that the
levels of IL-1β in the blood serum and in the cerebrospinal fluid
are increased in patients with significantly more stressful lives
and in patients who suffer from depression (78,79).
Furthermore, the expression levels of IL-1β in peripheral blood
circulation of patients with depression is correlated with the
severity and duration of depression (77).
TNF-α in depression
Chronic illnesses induce the release of peripheral
cytokines, which may result in cerebral endothelial leakage and
then cause a neuroinflammatory reaction (80). The induced inflammatory response in
the CNS is associated with the development of depression (81). TNF-α levels may be increased in
patients who suffer from depression and is a potential biomarker
for diagnosis of depression (82).
In a pre-clinical study based on an animal model, deletion of TNF-α
receptors was associated with antidepressant-like effects in
behavioural tests compared with the wild-type counterparts
(83). Additionally, a previous
study showed that inhibiting the TNF-α signaling pathway may serve
as a novel means of investigating how the CNS inflammatory response
is associated with depression (81).
Compounds targeting TNF-α signaling through inhibition of its
downstream effectors exert anti-depressant and anti-anxiety-like
effects in an animal model (81).
Other cytokines involved in
depression
Interestingly, a recent study found that the
antidepressant venlafaxine could ameliorate arthritis by targeting
STAT3 and IL17(84). Clinical data
has validated that IFN-γ levels are well correlated with clinical
symptoms of depression in patients with glioma (85). Although there is no effort to explore
whether IFN-γ activated the STAT cascade in depression, data have
shown that IFN-α can activate JNK/STATs in a model of depression
(86).
6. Conclusion
The pathogenesis of RA is complicated. It involves
several immune cells and the interplay between numerous cytokines
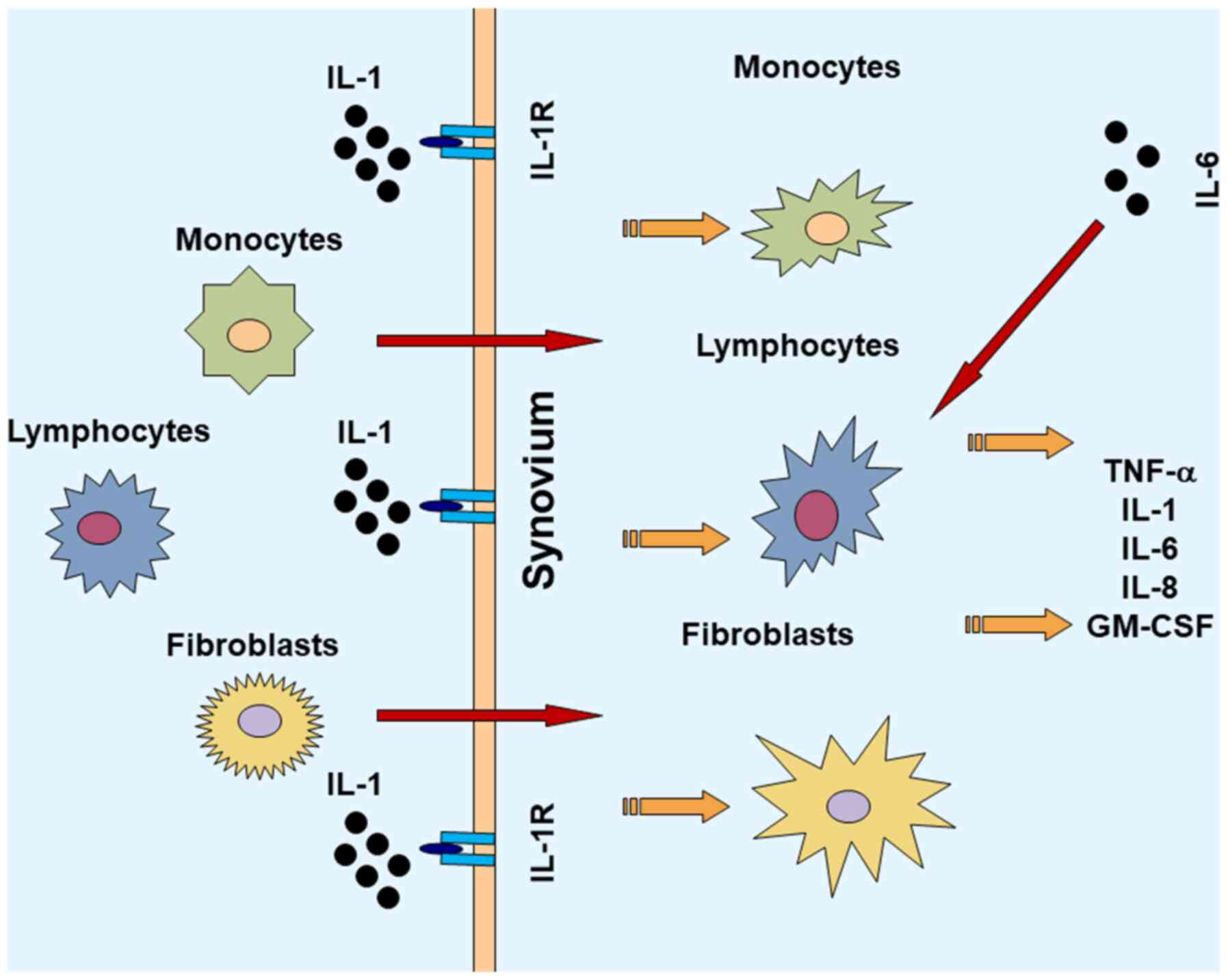
(Fig. 1) (87,88).
Consequently, these cytokines further activate and promote
differentiation of the downstream cells, which could result in
local or systemic symptoms relevant to RA. For example, certain
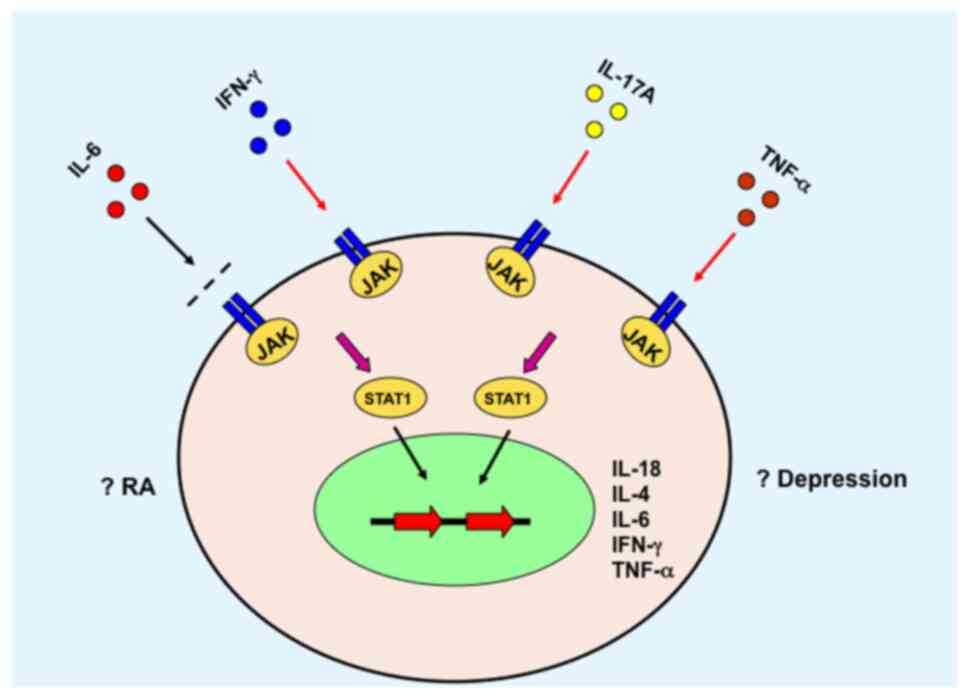
cytokines activate the JAK/STAT signaling pathway and initiate gene
transcription of various other cytokines (Fig. 2) (89,90),
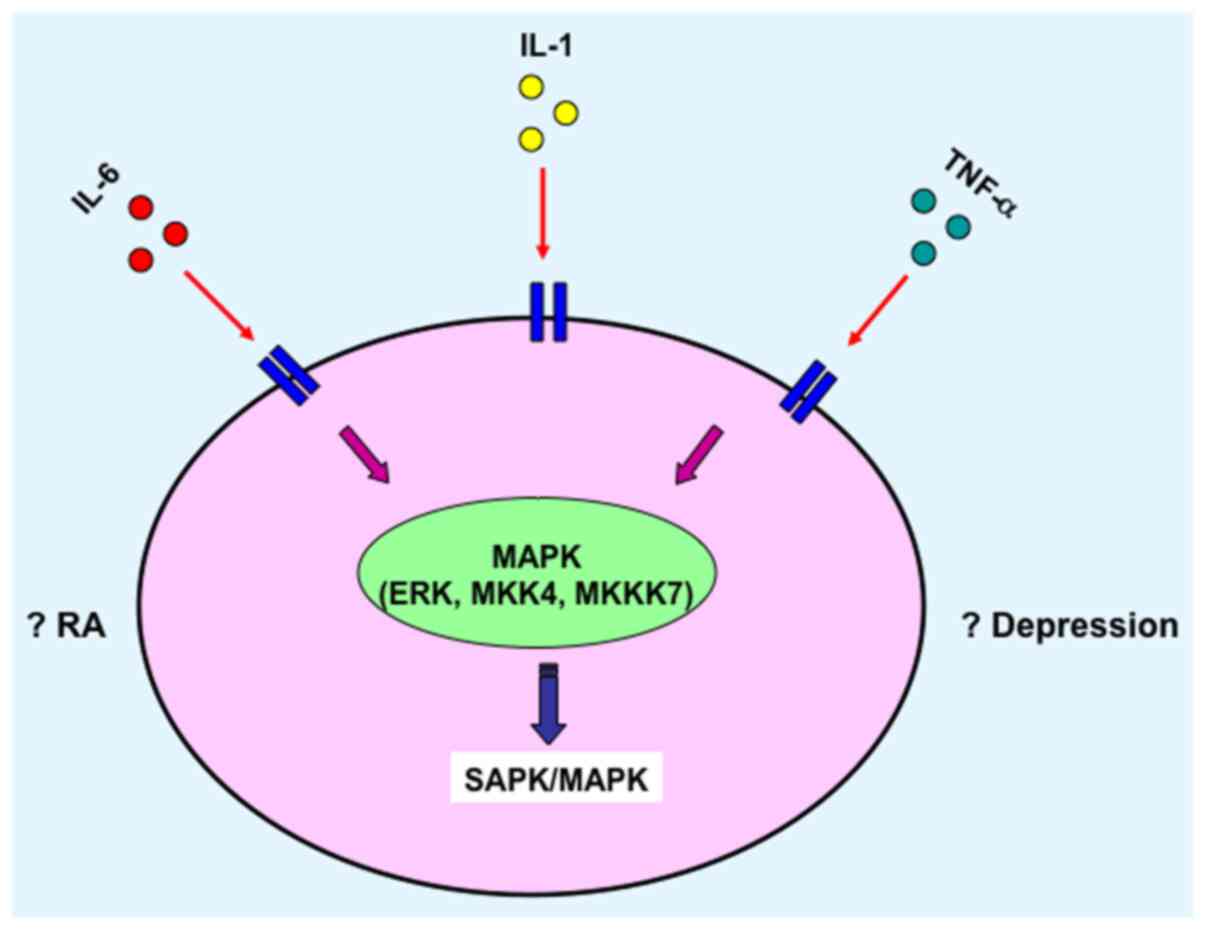
and/or activate the SAPK/MAPK (Fig.
3) (91,92). Furthermore, depression is a
frequently observed complication in patients with later stage RA.
These two diseases may aggravate each other in via unknown
mechanisms and afflict the patients further. This review focused on
the inflammatory cytokines and signaling pathways which may serve
as potential mediators of common pathways between RA and
depression. One of the purposes of this review was to identify
potential common therapeutic targets for both RA and depression
treatment. With the encouraging preclinical and clinical data,
there may be progress in therapeutic advancement targeting the
unresolved clinical hurdles in RA with depression. To further
elucidate the role of inflammation on the pathology of these
diseases, and to design improved treatments, comprehensive analyses
of mechanisms of action are necessary to optimise therapy with the
ultimate focus of providing a cure for patients with these chronic
illnesses. Additionally, RA is a chronic disease that is
potentially devastating for a patient's quality of life and may
result in mental disorders. Therefore, psychological factors may
also be another key network responsible for the depression and
other mood disorders in patients with RA.
Acknowledgements
Not applicable.
Funding
This review was supported by a Fellowship from China
Scholarship Council for the support of his overseas research at the
University of Manitoba (Canada).
Availability of data and materials
Not applicable.
Authors' contributions
CZ wrote and revised the manuscript. The author read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brennan FM and McInnes IB: Evidence that
cytokines play a role in rheumatoid arthritis. J Clin Invest.
118:3537–3545. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Ungethuem U, Haeupl T, Witt H, Koczan D,
Krenn V, Huber H, von Helversen TM, Drungowski M, Seyfert C, Zacher
J, et al: Molecular signatures and new candidates to target the
pathogenesis of rheumatoid arthritis. Physiol Genomics.
42A:267–282. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nixon R, Bansback N and Brennan A: The
efficacy of inhibiting tumour necrosis factor alpha and interleukin
1 in patients with rheumatoid arthritis: A meta-analysis and
adjusted indirect comparisons. Rheumatology (Oxford). 46:1140–1147.
2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Verhoef LM, van den Bemt BJ, van der Maas
A, Vriezekolk JE, Hulscher ME, van den Hoogen FH, Jacobs WC, van
Herwaarden N and den Broeder AA: Down-titration and discontinuation
strategies of tumour necrosis factor-blocking agents for rheumatoid
arthritis in patients with low disease activity. Cochrane Database
Syst Rev. 5(CD010455)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Upchurch KS and Kay J: Evolution of
treatment for rheumatoid arthritis. Rheumatology (Oxford).
51:vi28–vi36. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Quan LD, Thiele GM, Tian J and Wang D: The
development of novel therapies for rheumatoid arthritis. Expert
Opin Ther Pat. 18:723–738. 2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
O'Dell JR: Therapeutic strategies for
rheumatoid arthritis. N Engl J Med. 350:2591–2602. 2004.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mori S: Management of rheumatoid arthritis
patients with interstitial lung disease: Safety of biological
antirheumatic drugs and assessment of pulmonary fibrosis. Clin Med
Insights Circ Respir Pulm Med. 9 (Suppl 1):S41–S49. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Klarenbeek NB, Güler-Yüksel M, van der
Kooij SM, Han KH, Ronday HK, Kerstens PJ, Seys PE, Huizinga TW,
Dijkmans BA and Allaart CF: The impact of four dynamic,
goal-steered treatment strategies on the 5-year outcomes of
rheumatoid arthritis patients in the BeSt study. Ann Rheum Dis.
70:1039–1046. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Inui K and Koike T: Combination therapy
with biologic agents in rheumatic diseases: Current and future
prospects. Ther Adv Musculoskelet Dis. 8:192–202. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bruce TO: Comorbid depression in
rheumatoid arthritis: Pathophysiology and clinical implications.
Curr Psychiatry Rep. 10:258–264. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jacob L, Rockel T and Kostev K: Depression
risk in patients with rheumatoid arthritis in the United Kingdom.
Rheumatol Ther. 4:195–200. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Margaretten M, Julian L, Katz P and Yelin
E: Depression in patients with rheumatoid arthritis: Description,
causes and mechanisms. Int J Clin Rheumtol. 6:617–623.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Schiepers OJ, Wichers MC and Maes M:
Cytokines and major depression. Prog Neuropsychopharmacol Biol
Psychiatry. 29:201–217. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Viatte S, Plant D and Raychaudhuri S:
Genetics and epigenetics of rheumatoid arthritis. Nat Rev
Rheumatol. 9:141–153. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McInnes IB and Schett G: Cytokines in the
pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 7:429–442.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Firestein GS: Evolving concepts of
rheumatoid arthritis. Nature. 423:356–361. 2003.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kinne RW, Stuhlmüller B and Burmester GR:
Cells of the synovium in rheumatoid arthritis. Macrophages.
Arthritis Res Ther. 9(224)2007.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Kinne RW, Bräuer R, Stuhlmüller B,
Palombo-Kinne E and Burmester GR: Macrophages in rheumatoid
arthritis. Arthritis Res. 2:189–202. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Hata H, Sakaguchi N, Yoshitomi H, Iwakura
Y, Sekikawa K, Azuma Y, Kanai C, Moriizumi E, Nomura T, Nakamura T
and Sakaguchi S: Distinct contribution of IL-6, TNF-alpha, IL-1,
and IL-10 to T cell-mediated spontaneous autoimmune arthritis in
mice. J Clin Invest. 114:582–588. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Schett G and Gravallese E: Bone erosion in
rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat Rev
Rheumatol. 8:656–664. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Silveira KD, Coelho FM, Vieira AT, Barroso
LC, Queiroz-Junior CM, Costa VV, Sousa LF, Oliveira ML, Bader M,
Silva TA, et al: Mechanisms of the anti-inflammatory actions of the
angiotensin type 1 receptor antagonist losartan in experimental
models of arthritis. Peptides. 46:53–63. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ruscitti P, Cipriani P, Liakouli V,
Carubbi F, Berardicurti O, Di Benedetto P, Ciccia F, Guggino G,
Alvaro S, Triolo G and Giacomelli R: The emerging role of IL-1
inhibition in patients affected by rheumatoid arthritis and
diabetes. Rev Recent Clin Trials. 13:210–214. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi
M, Zhan Z, Liang L, Yang X and Xu H: Inhibition of
6-phosphofructo-2-kinase suppresses fibroblast-like
synoviocytes-mediated synovial inflammation and joint destruction
in rheumatoid arthritis. Br J Pharmacol. 174:893–908.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Buckley CD: Michael Mason prize essay
2003. Why do leucocytes accumulate within chronically inflamed
joints? Rheumatology (Oxford). 42:1433–1444. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kay J and Calabrese L: The role of
interleukin-1 in the pathogenesis of rheumatoid arthritis.
Rheumatology (Oxford). 43 (Suppl 3):iii2–iii9. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Arend WP, Palmer G and Gabay C: IL-1,
IL-18, and IL-33 families of cytokines. Immunol Rev. 223:20–38.
2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nakae S, Saijo S, Horai R, Sudo K, Mori S
and Iwakura Y: IL-17 production from activated T cells is required
for the spontaneous development of destructive arthritis in mice
deficient in IL-1 receptor antagonist. Proc Natl Acad Sci USA.
100:5986–5990. 2003.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Abramson SB and Amin A: Blocking the
effects of IL-1 in rheumatoid arthritis protects bone and
cartilage. Rheumatology (Oxford). 41:972–980. 2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jacques C, Gosset M, Berenbaum F and Gabay
C: The role of IL-1 and IL-1Ra in joint inflammation and cartilage
degradation. Vitam Horm. 74:371–403. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lebre MC, Jongbloed SL, Tas SW, Smeets TJ,
McInnes IB and Tak PP: Rheumatoid arthritis synovium contains two
subsets of CD83-DC-LAMP-dendritic cells with distinct cytokine
profiles. Am J Pathol. 172:940–950. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Plater-Zyberk C, Joosten LA, Helsen MM,
Sattonnet-Roche P, Siegfried C, Alouani S, van De Loo FA, Graber P,
Aloni S, Cirillo R, et al: Therapeutic effect of neutralizing
endogenous IL-18 activity in the collagen-induced model of
arthritis. J Clin Invest. 108:1825–1832. 2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lotz M: The role of nitric oxide in
articular cartilage damage. Rheum Dis Clin North Am. 25:269–282.
1999.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tanaka T, Narazaki M and Kishimoto T: IL-6
in inflammation, immunity, and disease. Cold Spring Harb Perspect
Biol. 6(a016295)2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Alonzi T, Fattori E, Lazzaro D, Costa P,
Probert L, Kollias G, De Benedetti F, Poli V and Ciliberto G:
Interleukin 6 is required for the development of collagen-induced
arthritis. J Exp Med. 187:461–468. 1998.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kishimoto T: Interleukin-6: From basic
science to medicine-40 years in immunology. Annu Rev Immunol.
23:1–21. 2005.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Narazaki M, Tanaka T and Kishimoto T: The
role and therapeutic targeting of IL-6 in rheumatoid arthritis.
Expert Rev Clin Immunol. 13:535–551. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Marrelli A, Cipriani P, Liakouli V,
Carubbi F, Perricone C, Perricone R and Giacomelli R: Angiogenesis
in rheumatoid arthritis: A disease specific process or a common
response to chronic inflammation? Autoimmun Rev. 10:595–598.
2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Johnson KE and Wilgus TA: Vascular
endothelial growth factor and angiogenesis in the regulation of
cutaneous wound repair. Adv Wound Care (New Rochelle). 3:647–661.
2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sokolove J and Lepus CM: Role of
inflammation in the pathogenesis of osteoarthritis: Latest findings
and interpretations. Ther Adv Musculoskelet Dis. 5:77–94.
2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Diaz-Torne C, Ortiz MDA, Moya P, Hernandez
MV, Reina D, Castellvi I, De Agustin JJ, Fuente D, Corominas H,
Sanmarti R, et al: The combination of IL-6 and its soluble receptor
is associated with the response of rheumatoid arthritis patients to
tocilizumab. Semin Arthritis Rheum. 47:757–764. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Aggarwal BB: Signalling pathways of the
TNF superfamily: A double-edged sword. Nat Rev Immunol. 3:745–756.
2003.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Holnthoner W, Bonstingl C, Hromada C,
Muehleder S, Zipperle J, Stojkovic S, Redl H, Wojta J, Schöchl H,
Grillari J, et al: Endothelial cell-derived extracellular vesicles
size-dependently exert procoagulant activity detected by
thromboelastometry. Sci Rep. 7(3707)2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Horiuchi T, Mitoma H, Harashima S,
Tsukamoto H and Shimoda T: Transmembrane TNF-alpha: Structure,
function and interaction with anti-TNF agents. Rheumatology
(Oxford). 49:1215–1228. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chen X and Oppenheim JJ: Contrasting
effects of TNF and anti-TNF on the activation of effector T cells
and regulatory T cells in autoimmunity. FEBS Lett. 585:3611–3618.
2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yamagishi S, Ohnishi M and Pawankar R:
IL-1 and TNF-alpha-mediated regulation of IL-6, IL-8, and GM-CSF
release from cultured nasal epithelial cells. Nihon Jibiinkoka
Gakkai Kaiho. 103:829–835. 2000.PubMed/NCBI View Article : Google Scholar : (In Japanese).
|
|
47
|
Fiocco U, Sfriso P, Oliviero F, Lunardi F,
Calabrese F, Scagliori E, Cozzi L, Di Maggio A, Nardacchione R,
Molena B, et al: Blockade of intra-articular TNF in peripheral
spondyloarthritis: Its relevance to clinical scores, quantitative
imaging and synovial fluid and synovial tissue biomarkers. Joint
Bone Spine. 80:165–170. 2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Bevaart L, Vervoordeldonk MJ and Tak PP:
Evaluation of therapeutic targets in animal models of arthritis:
How does it relate to rheumatoid arthritis? Arthritis Rheum.
62:2192–2205. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Monach PA, Benoist C and Mathis D: The
role of antibodies in mouse models of rheumatoid arthritis, and
relevance to human disease. Adv Immunol. 82:217–248.
2004.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Hassan S, Milman U, Feld J, Eder L, Lavi
I, Cohen S and Zisman D: Effects of anti-TNF-α treatment on lipid
profile in rheumatic diseases: An analytical cohort study.
Arthritis Res Ther. 18(261)2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ma X and Xu S: TNF inhibitor therapy for
rheumatoid arthritis. Biomed Rep. 1:177–184. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bourne T, Fossati G and Nesbitt A: A
PEGylated Fab' fragment against tumor necrosis factor for the
treatment of Crohn disease: Exploring a new mechanism of action.
BioDrugs. 22:331–337. 2008.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Capria A, De Nardo D, Baffetti FR, Barbini
U, Violo A, Tondo T and Fontana L: Long-term anti-TNF-alpha
treatments reverse the endothelial dysfunction in rheumatoid
arthritis: The biological coherence between synovial and
endothelial inflammation. Int J Immunopathol Pharmacol. 23:255–262.
2010.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Tam LS, Kitas GD and Gonzalez-Gay MA: Can
suppression of inflammation by anti-TNF prevent progression of
subclinical atherosclerosis in inflammatory arthritis? Rheumatology
(Oxford). 53:1108–1119. 2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Moelants EA, Mortier A, Van Damme J and
Proost P: Regulation of TNF-α with a focus on rheumatoid arthritis.
Immunol Cell Biol. 91:393–401. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lis K, Kuzawińska O and Balkowiec-Iskra E:
Tumor necrosis factor inhibitors-state of knowledge. Arch Med Sci.
10:1175–1185. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Radner H and Aletaha D: Anti-TNF in
rheumatoid arthritis: An overview. Wien Med Wochenschr. 165:3–9.
2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
van Schouwenburg PA, Rispens T and Wolbink
GJ: Immunogenicity of anti-TNF biologic therapies for rheumatoid
arthritis. Nat Rev Rheumatol. 9:164–172. 2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Dommasch E and Gelfand JM: Is there truly
a risk of lymphoma from biologic therapies? Dermatol Ther.
22:418–430. 2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wolfe F and Michaud K: Biologic treatment
of rheumatoid arthritis and the risk of malignancy: Analyses from a
large US observational study. Arthritis Rheum. 56:2886–2895.
2007.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Bullock J, Rizvi SAA, Saleh AM, Ahmed SS,
Do DP, Ansari RA and Ahmed J: Rheumatoid arthritis: A brief
overview of the treatment. Med Princ Pract. 27:501–507.
2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Kalliolias GD and Liossis SN: The future
of the IL-1 receptor antagonist anakinra: From rheumatoid arthritis
to adult-onset Still's disease and systemic-onset juvenile
idiopathic arthritis. Expert Opin Investig Drugs. 17:349–359.
2008.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ramírez J and Cañete JD: Anakinra for the
treatment of rheumatoid arthritis: A safety evaluation. Expert Opin
Drug Saf. 17:727–732. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Bresnihan B, Newmark R, Robbins S and
Genant HK: Effects of anakinra monotherapy on joint damage in
patients with rheumatoid arthritis. Extension of a 24-week
randomized, placebo-controlled trial. J Rheumatol. 31:1103–1111.
2004.PubMed/NCBI
|
|
65
|
Araki M, Matsuoka T, Miyamoto K, Kusunoki
S, Okamoto T, Murata M, Miyake S, Aranami T and Yamamura T:
Efficacy of the anti-IL-6 receptor antibody tocilizumab in
neuromyelitis optica: A pilot study. Neurology. 82:1302–1306.
2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Scott LJ: Tocilizumab: A review in
rheumatoid arthritis. Drugs. 77:1865–1879. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Smolen JS, Beaulieu A, Rubbert-Roth A,
Ramos-Remus C, Rovensky J, Alecock E, Woodworth T and Alten R:
OPTION Investigators. Effect of interleukin-6 receptor inhibition
with tocilizumab in patients with rheumatoid arthritis (OPTION
study): A double-blind, placebo controlled, randomised trial.
Lancet. 371:987–997. 2008.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Maini RN, Taylor PC, Szechinski J, Pavelka
K, Bröll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, et
al: Double-blind randomized controlled clinical trial of the
interleukin-6 receptor antagonist, tocilizumab, in European
patients with rheumatoid arthritis who had an incomplete response
to methotrexate. Arthritis Rheum. 54:2817–2829. 2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Yip RML and Yim CW: Role of interleukin-6
inhibitors in the management of rheumatoid arthritis. J Clin
Rheumatol:. 2019, doi: 10.1097/RHU.0000000000001293 (Online ahead
of print).
|
|
70
|
Park JY and Pillinger MH: Interleukin-6 in
the pathogenesis of rheumatoid arthritis. Bull NYU Hosp Jt Dis. 65
(Suppl 1):S4–S10. 2007.PubMed/NCBI
|
|
71
|
Conigliaro P, Triggianese P, De Martino E,
Fonti GL, Chimenti MS, Sunzini F, Viola A, Canofari C and Perricone
R: Challenges in the treatment of rheumatoid arthritis. Autoimmun
Rev. 18:706–713. 2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Emery P, Pope JE, Kruger K, Lippe R,
DeMasi R, Lula S and Kola B: Efficacy of monotherapy with biologics
and JAK inhibitors for the treatment of rheumatoid arthritis: A
systematic review. Adv Ther. 35:1535–1563. 2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Gaffen SL: The role of interleukin-17 in
the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep.
11:365–370. 2009.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Kasperkovitz PV, Verbeet NL, Smeets TJ,
van Rietschoten JG, Kraan MC, van der Pouw Kraan TC, Tak PP and
Verweij CL: Activation of the STAT1 pathway in rheumatoid
arthritis. Ann Rheum Dis. 63:233–239. 2004.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Hodes GE, Ménard C and Russo SJ:
Integrating Interleukin-6 into depression diagnosis and treatment.
Neurobiol Stress. 4:15–22. 2016.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Khandaker GM, Pearson RM, Zammit S, Lewis
G and Jones PB: Association of serum interleukin 6 and C-reactive
protein in childhood with depression and psychosis in young adult
life: A population-based longitudinal study. JAMA Psychiatry.
71:1121–1128. 2014.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Koo JW and Duman RS: Evidence for IL-1
receptor blockade as a therapeutic strategy for the treatment of
depression. Curr Opin Investig Drugs. 10:664–671. 2009.PubMed/NCBI
|
|
78
|
Raison CL, Capuron L and Miller AH:
Cytokines sing the blues: Inflammation and the pathogenesis of
depression. Trends Immunol. 27:24–31. 2006.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Steptoe A, Hamer M and Chida Y: The
effects of acute psychological stress on circulating inflammatory
factors in humans: A review and meta-analysis. Brain Behav Immun.
21:901–912. 2007.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Liu H, Luiten PG, Eisel UL, Dejongste MJ
and Schoemaker RG: Depression after myocardial infarction:
TNF-α-induced alterations of the blood-brain barrier and its
putative therapeutic implications. Neurosci Biobehav Rev.
37:561–572. 2013.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Abbott R, Whear R, Nikolaou V, Bethel A,
Coon JT, Stein K and Dickens C: Tumour necrosis factor-α inhibitor
therapy in chronic physical illness: A systematic review and
meta-analysis of the effect on depression and anxiety. J Psychosom
Res. 79:175–184. 2015.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Kamel KM, Gad AM, Mansour SM, Safar MM and
Fawzy HM: Venlafaxine alleviates complete Freund's adjuvant-induced
arthritis in rats: Modulation of STAT-3/IL-17/RANKL axis. Life Sci.
226:68–76. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Song L, Quan X, Su L, Wang K, Wang H, Wu
L, Chen C, Li S, Xiang W, Chen L and Zhou J: Inflammation and
behavioral symptoms in preoperational glioma patients: Is
depression, anxiety, and cognitive impairment related to markers of
systemic inflammation? Brain Behav. 10(e01771)2020.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Lu DY, Leung YM and Su LP: Interferon-α
induces nitric oxide synthase expression and haem oxygenase-1
down-regulation in microglia: Implications of cellular mechanism of
IFN-α-induced depression. Int J Neuropsychopharmacol. 16:433–444.
2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Carboni l, McCarthy DJ, Delafont B, Filosi
M, Ivanchenko E, Ratti E, Learned SM, Alexander R and Domenici E:
Biomarkers for response in major depression: Comparing paroxetine
and venlafaxine from two randomised placebo-controlled clinical
studies. Transl Psychiatry. 9(182)2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Simen BB, Duman CH, Simen AA and Duman RS:
TNFalpha signaling in depression and anxiety: Behavioral
consequences of individual receptor targeting. Biol Psychiatry.
59:775–785. 2006.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Telfer JF and Brock JH: Proinflammatory
cytokines increase iron uptake into human monocytes and synovial
fibroblasts from patients with rheumatoid arthritis. Med Sci Monit.
10:BR91–BR95. 2004.PubMed/NCBI
|
|
88
|
Jenkins JK, Hardy KJ and McMurray RW: The
pathogenesis of rheumatoid arthritis: A guide to therapy. Am J Med
Sci. 323:171–180. 2002.PubMed/NCBI View Article : Google Scholar
|
|
89
|
De Simone V, Franzè E, Ronchetti G,
Colantoni A, Fantini MC, Di Fusco D, Sica GS, Sileri P, MacDonald
TT, Pallone F, et al: Th17-type cytokines, IL-6 and TNF-α
synergistically activate STAT3 and NF-kB to promote colorectal
cancer cell growth. Oncogene. 34:3493–3503. 2015.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Kato H, Endres J and Fox DA: The roles of
IFN-γ versus IL-17 in pathogenic effects of human Th17 cells on
synovial fibroblasts. Mod Rheumatol. 23:1140–1150. 2013.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Campbell J, Ciesielski CJ, Hunt AE,
Horwood NJ, Beech JT, Hayes LA, Denys A, Feldmann M, Brennan FM and
Foxwell BM: A novel mechanism for TNF-alpha regulation by p38 MAPK:
Involvement of NF-kappa B with implications for therapy in
rheumatoid arthritis. J Immunol. 173:6928–6937. 2004.PubMed/NCBI View Article : Google Scholar
|
|
92
|
De Cesaris P, Starace D, Riccioli A,
Padula F, Filippini A and Ziparo E: Tumor necrosis factor-alpha
induces interleukin-6 production and integrin ligand expression by
distinct transduction pathways. J Biol Chem. 273:7566–5671.
1998.PubMed/NCBI View Article : Google Scholar
|