Introduction
Damage-associated molecular patterns (DAMPs) are
associated with inflammatory diseases such as sepsis, rheumatoid
arthritis (RA), atherosclerosis, cerebral infarction and
periodontitis (1-4).
In general, in living cells, the presence of DAMPs in the
intracellular space is physiologically normal and are not harmful,
as they contribute to processes associated with cell maintenance,
such as cell cycle progression, DNA construction and gene
expression (5-7).
However, proteins released by damaged or necrotic cells, including
DAMPs, can be dangerously proinflammatory, causing cytotoxicity to
living cells (5-7).
DAMPs include cellular molecules, such as the nuclear proteins,
high mobility group box 1 (HMGB1), histones H3 and H4, and
nucleophosmin 1 (NPM1) (1-6).
The involvement of HMGB1 was first demonstrated in a patient with
sepsis (7). Histones H3 and H4
induce platelet activation and endothelial cell death (6). Both NPM1 and HMGB1 induce the
production of inflammatory cytokines, such as tumor necrosis factor
(TNF)-α, interleukin (IL)-6 and IL-8. DAMPs negatively influence
the prognosis of inflammatory diseases (1-7).
As their activity may have fatal consequences for patients with
inflammatory diseases (6,7), regulating DAMPs is essential.
In our previous study, it was shown that the
ubiquitously expressed nucleolar phosphoprotein, NPM1 (also known
as B23, numatrin and NO38) (8), was
a novel DAMP (5). Intracellular NPM1
regulates ribosome biogenesis, the response to genotoxic stress and
the inhibition of hypoxia-induced apoptosis (8), thus NPM1 also contributes to cell
maintenance. NPM1, which is more abundant in tumor cells than in
normal resting cells, shuttles between the nucleus and cytoplasm
during the cell cycle (8,9). Thus, NPM1 is an essential molecule in
living cells.
NPM1 has been shown to be released from damaged or
activated murine macrophage-like RAW264.7 cells (5). Extracellular NPM1 acts as an
inflammatory cytokine by inducing the production of the
inflammatory cytokine, TNF-α, via ERK-1/2 activation in RAW264.7
cells, but not via the kinases, c- JNK and p38 MAPK (5). Furthermore, in the sepsis model, cecal
ligation and puncture (CLP), NPM1 was detected in serum derived
from model rats, but not in serum from control rats (5). Thus, in the extracellular space, NPM1
may act as a cytokine as well as a novel DAMP. However, the NPM1
receptor on the cell membrane has not yet been identified.
The ligands of 9 out of 10 human homologs of
Toll-like receptors (TLRs) have been identified as DAMPs (10-12).
TLR4 was first identified as a DAMP receptor, and is the most
extensively studied receptor amongst the TLR family. TLR4
associated with myeloid differentiation protein (MD)-2 (TLR4/MD-2)
is a receptor for lipopolysaccharide (LPS), which is a component of
the outer membrane of Gram-negative bacteria (13). The effects of LPS are mediated via
TLR4/MD-2 expressed by endothelial cells as well as immune cells
(macrophages and dendritic cells). Apart from LPS, TLR4 is
activated by endogenous molecules, including DAMPs, such as HMGB1,
histone H3 and histone H4 (10-12).
In the TLR4/MD-2 signal transduction pathway, TLR4/MD-2 dimerizes
with another TLR4/MD-2 and recruits specific intracellular adaptor
molecules to promote the activation of downstream signaling
pathways. These pathways include the myeloid differentiation
primary response gene 88 (MyD88)-dependent pathway and the
MyD88-independent pathway, which includes Toll/IL-1 receptor
(TIR)-domain containing adaptor-inducing interferon-β (TRIF). Both
pathways activate nuclear factor-κB (NF-κB) signaling, but only the
TRIF pathway stimulates signaling by interferon regulatory factor
3. These pathways may induce the production of inflammatory
cytokines, such as TNF-α, IL-6 and IL-8(13). Thus, identifying TLR4/MD-2 ligands
may be a novel strategy for treating inflammatory diseases, such as
sepsis, cancer and RA. Indeed, molecules targeting TLR4/MD-2 have
been developed; for example, LPS-RS as an MD-2 antagonist and
TAK-242 as an intracellular signaling inhibitor of TLR4 (14-17).
In the present study, whether TLR4/MD-2 was the
receptor for extracellular NPM1 was assessed. To this end, a
reporter gene assay with TLR4/MD-2-expressing cells and control 293
cells was used, and TNF-α production was measured using phorbol
12-myristate 13-acetate (PMA)-differentiated THP-1 cells.
Furthermore, using LPS-RS and TAK-242, whether TNF-α production
upon NPM1 stimulation was mediated via TLR4/MD-2, and whether
stimulation activated the TLR4/MD-2/ERK1/2 signaling pathway, was
investigated. Additionally, far-western blotting was used to
examine direct binding of NPM1 to MD-2. The results showed that
NPM1 binds to MD-2 to induce TNF-α production.
Materials and methods
Reagents
Bacterial expression vectors (pGEX6p-1) encoding the
fusion proteins, GST-NPM1 or GST-MD-2, were purchased from
GenScript. These proteins were affinity-purified using
glutathione-Sepharose beads and cleaved by Turbo3C protease to
remove the GST from the indicated protein, according to the
manufacturer's protocol (Nacalai Tesque, Inc.). Unless indicated
otherwise, PMA and other reagents were purchased from Sigma-Aldrich
(Merck KGaA). LPS-Rhodobacter sphaeroides (RS) (TLR4
antagonist) and TAK-242 (a TLR4 signaling inhibitor) were purchased
from InvivoGen.
Cell culture
THP-1 cells were obtained from the American Type
Culture Collection and maintained in RPMI-1640 medium (Nacalai
Tesque, Inc.) supplemented with 10% FBS and 2 mM glutamine. A total
of 4x105 cells/ml were treated with 10 nM PMA for 16 h
(18), and then treated with
recombinant NPM1 (rNPM1), as indicated in the figures.
Reporter gene assay
To assess whether NPM1 activates the TLR4/MD-2
pathway, human TLR4/NF-κB/SEAP reporter 293 cells (Blue hTLR4
cells), in which the expression of the SEAP reporter gene is
controlled by an IL-12 p40 minimal promoter fused to five
NF-κB and AP-1-binding sites (InvivoGen) were used. Treating these
cells with a TLR4 ligand activates NF-κB and AP-1, which induces
the production of SEAP. In parallel, mock plasmid-transfected 293
cells (Null) was used as a TLR4-negative control. Briefly, 50 ng/ml
NPM1 (ATGen, Ltd.) was added to cultures of Blue hTLR4 cells or
Null cells for 16 h. TLR4 signaling was evaluated by measuring SEAP
activity at an optical density of 650 nm in Quanti-Blue reagent
(InvivoGen). Experiments were repeated three times.
Treatment with purified NPM1 or
heat-inactivated purified NPM1
THP-1 cells were incubated with 50 nM PMA for 24 h
and then washed with Opti-MEM (Thermo Fisher Scientific, Inc.). The
cells were then incubated with native NPM1 or heat-inactivated NPM1
(100˚C for 5 min) at a final concentration of 5 nM for 16 h.
Experiments were repeated nine times.
Effects of rNPM1 in the presence or
absence of LPS-RS or TAK-242
THP-1 cells were incubated with 50 nM PMA for 24 h
and then washed with Opti-MEM (Thermo Fisher Scientific, Inc.). The
cells were then incubated with or without the TLR4 inhibitor,
LPS-RS or TAK-242. NPM1 was subsequently added to the cells (final
concentration, 5 nM) for 16 h. Experiments were repeated nine
times.
Detection of ERK1/2
phosphorylation
THP-1 cells were treated with PMA for 24 h, washed
and then incubated with 100 ng/ml LPS-RS or 100 nM TAK-242 for 2 h,
based on a previous study (5).
Subsequently, NPM1 was added to the cells (final concentration, 5
nM) for 30 min. Protein concentrations was determined using a
Bradford assay (Bio Rad Laboratories, Inc.). The treated cells were
then washed with cold PBS, and 200 µl cell lysis buffer containing
62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol and 0.002%
bromophenol blue were added. A total of 2 µg of the above proteins
in the resultant lysates (200 µl) were loaded on a SDS-gel,
resolved using SDS-PAGE and transferred to a nitrocellulose
membrane (GE Healthcare). After blocking with Block One (Nacalai
Tesque, Inc.) for 1 h at room temperature, the membrane was
incubated with anti-phospho (p)-ERK1/2 (1:1,000; cat. no. 4370S) or
anti-total (t)-ERK1/2 antibodies (Cell Signaling Technology, Inc.;
1:1,000; cat. no. 9102S) at 4˚C for 16 h. Subsequently, the
membranes were incubated with horse radish peroxidase
(HRP)-conjugated anti-rabbit antibodies (Santa Cruz Biotechnology
Inc.; cat. no. sc-2357) for 1 h at room temperature. After washing,
signals were detected using ImmunoStar Zeta reagent (FUJIFILM) and
measured using Image J version 1.52a (National Institutes of
Health).
Determination of cytokine
production
ELISA kits (BioLegend, Inc.; cat. no. 430201) were
used to quantify the TNF-α concentration in cell-free supernatants,
according to the manufacturer's protocol. Absorbance at 450 nm was
measured using a microplate reader. The sensitivity of the
commercial ELISA kit was 15.6 pg/ml for TNF-α.
Coomassie Brilliant Blue (CBB)
staining
GST-MD-2 induced using
isopropyl-β-D-thiogalactopyranoside (Nacalai Tesque, Inc.) was
concentrated using Centrifugal Filter Units (10,000 Da cut off; EMD
Millipore). Concentrated GST-MD-2 was mixed with an equal volume of
lysis buffer and boiled for 5 min. These samples (40 µl) were
loaded onto a 12% SDS-gel, resolved using SDS-PAGE and
subsequently, the gels were stained with CBB (Nacalai Tesque,
Inc.), as described previously (5).
Interaction between NPM1 and MD-2
The GST fusion proteins were expressed and purified
using glutathione-Sepharose beads (19). NPM1 was cleaved from GST bound to the
column using Turbo3C Protease (Nacalai Tesque, Inc.). NPM1 was
purified using LPS-free 0.9% NaCl. The interaction between NPM1 and
GST-MD-2 was measured as previously described (20). NPM1 (500 ng) was separated using a
15% SDS-gel and SDS-PAGE, and then transferred to a nitrocellulose
membrane. After blocking with Block One, the membrane was incubated
with 6 µg/ml GST-MD-2 or GST at 4˚C for 16 h. Subsequently, the
membrane was incubated with an anti-GST antibody (Medical &
Biological Laboratories Co., Ltd.; 1:1,000; cat. no. PM013-7) for 1
h at room temperature. After washing, the membrane was incubated
with HRP-conjugated anti-rabbit-IgG antibodies (Santa Cruz
Biotechnology, Inc.; 1:5,000; cat. no. sc-2357) for 1 h at room
temperature and the immune complexes were detected using ImmunoStar
Zeta reagent (FUJIFILM).
Statistical analysis
All statistical analysis was performed using
GraphPad version 8 (GraphPad Software, Inc.). Data are presented as
the mean ± the standard error of the mean. Differences between
groups were evaluated using a one-way ANOVA with a post-hoc Tukey's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
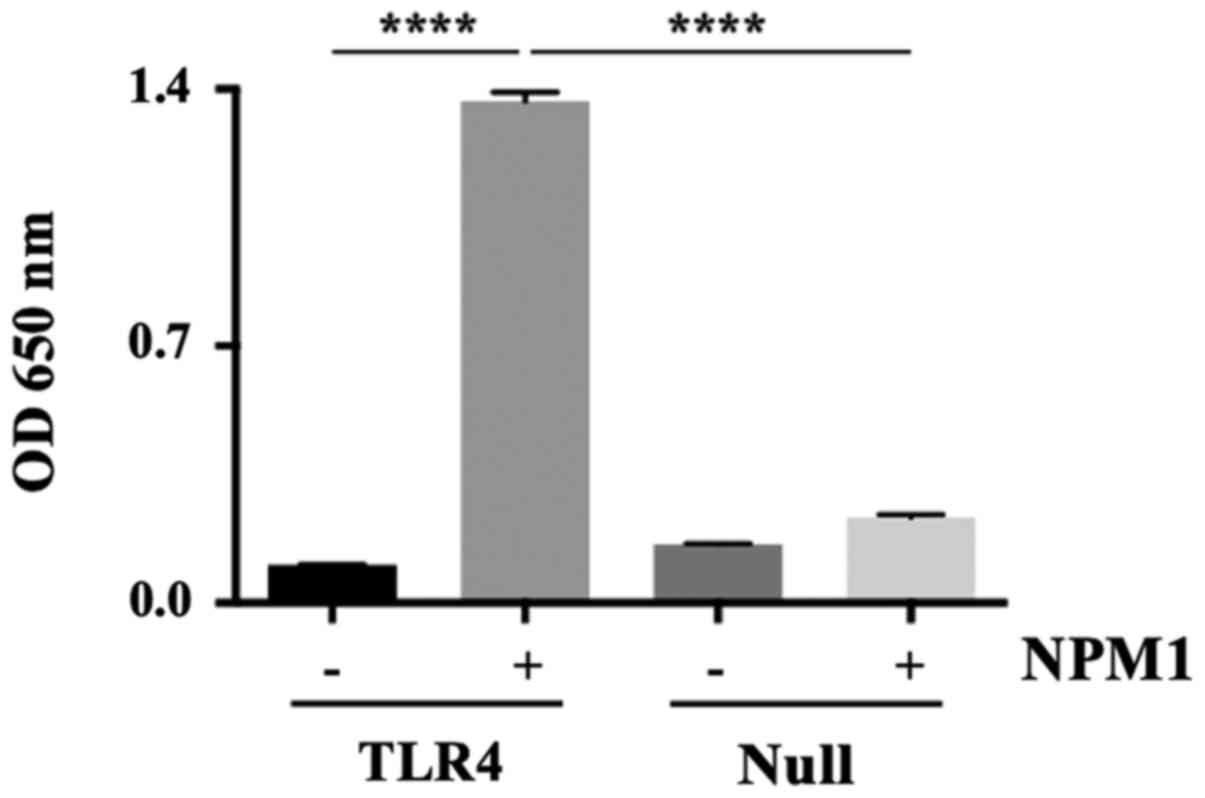
TLR4/MD-2 mediates the effects of
extracellular NPM1
To determine the effects of extracellular NPM1 on
293-TLR4-Blue cells and Null cells, culture supernatants were
assayed for SEAP activity. NPM1 significantly increased the SEAP
activity in 293-TLR4-Blue cells (13-fold increase; P<0.0001),
but not in the Null cells (Fig.
1).
TAK-242 and LPS-RS inhibits
NPM1-induced TNF-α production and ERK1/2 activation
Purified NPM1 was detected as a single band in
CBB-stained gels (Fig. 2A, left
CBB), corresponding to the specific band detected by the anti-NPM1
antibody (Fig. 2A, left western
blotting). Using the purified NPM1, whether NPM1 specifically
induced TNF-α production in PMA-differentiated THP-1 cells was next
investigated. As shown in Fig. 2A
(right), the native NPM1 significantly induced TNF-α production,
whereas the heated NPM1 did not.
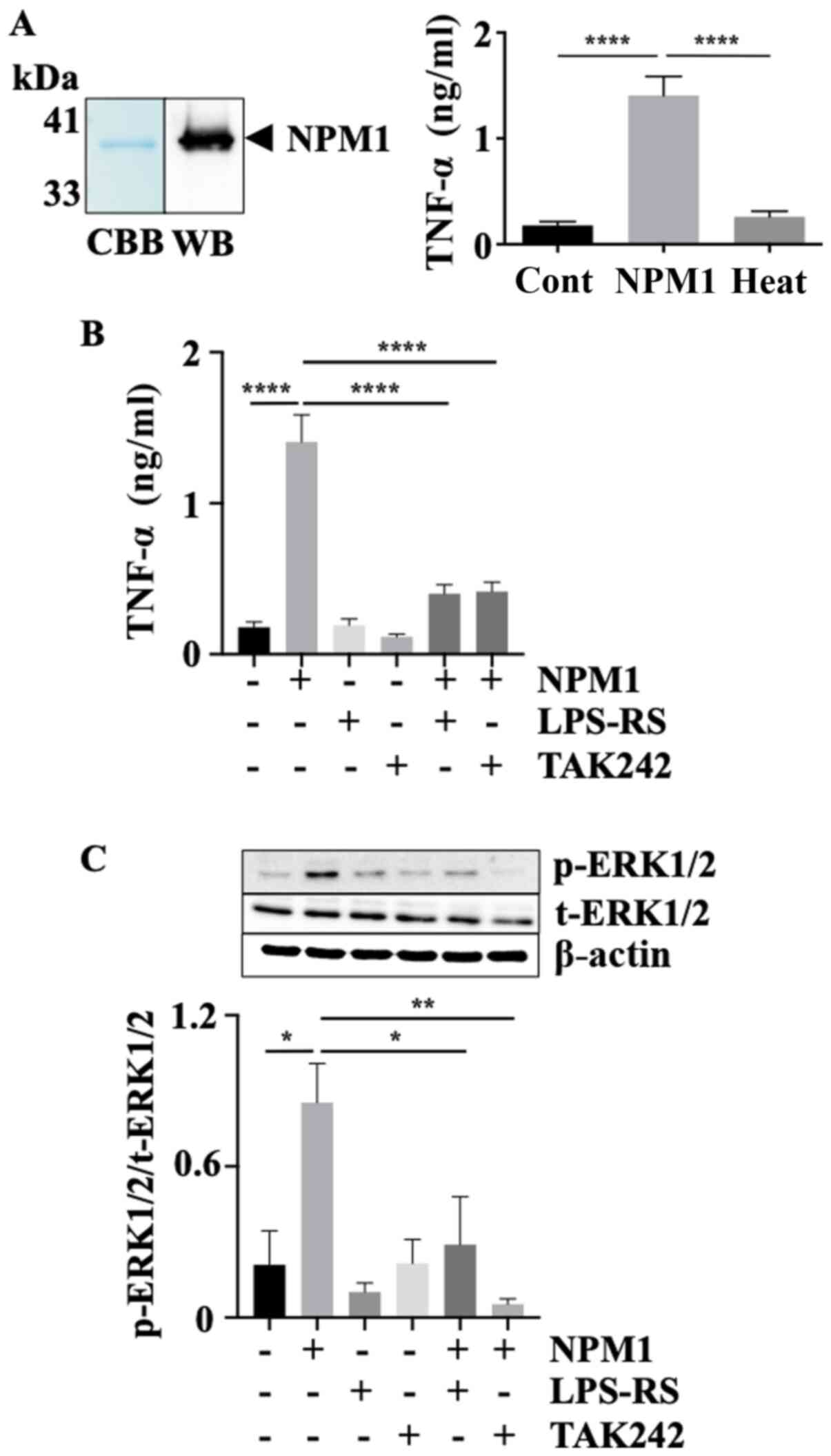 | Figure 2TAK-242 and LPS-RS inhibits NPM1
signaling. (A) SDS-PAGE and western blotting analysis of purified
recombinant NPM1 (left). PMA-differentiated THP-1 cells were
incubated with intact NPM1 or heat-inactivated NPM1. TNF-α levels
in the culture supernatants were determined (right). n=9 per group.
(B) TNF-α levels in culture supernatants of PMA-differentiated
THP-1 cells incubated with or without the MD-2 antagonist LPS-RS or
the TLR4 signaling inhibitor TAK-242, followed by addition of NPM1.
n=9 per group. (C) ERK1/2 phosphorylation levels in
PMA-differentiated THP-1 cells incubated with or without LPS-RS or
TAK-242 followed by addition of NPM1. n=4 per group. The upper
panel shows a representative western blotting of activated ERK1/2
(p-ERK1/2) and t-ERK1/2 following the indicated treatments with or
without NPM1, LPS-RS and/or TAK-242. β-actin was used as the
loading control. The lower panel shows quantitative analysis of the
p-ERK1/2/t-ERK1/2 expression ratio. Data are presented as the mean
± standard error of the mean of three independent experiments.
*P<0.05, **P<0.01,
****P<0.0001. NPM1, nucleophosmin 1; Heat,
heat-inactivated NPM1; PMA, phorbol 12-myristate 13-acetate; TLR,
toll-like receptor; MD-2, myeloid differentiation protein-2; p-
phospho; t-, total; CBB, Coomassie Brilliant Blue; WB, western
blotting. |
Subsequently, whether LPS-RS and TAK-242 inhibited
TNF-α production stimulated by 5 nM NPM1 in PMA-differentiated
THP-1 cells was assessed. NPM1 significantly induced TNF-α
production compared with the control (8-fold-increase;
P<0.0001), but not when the cells were pretreated with LPS-RS or
TAK-242 (Fig. 2B). In our previous
study, it was shown that TNF-α production upon NPM1 stimulation was
mediated via ERK1/2 activation, but not via other kinases, such as
JNK and p38 MAPK (5). Thus, whether
these two inhibitors prevented ERK1/2 activation by NPM1
stimulation was assessed. As shown by the p-ERK1/2 to t-ERK1/2
ratio in Fig. 2C, pretreatment with
LPS-RS or TAK-242 significantly suppressed the levels of
NPM1-induced p-ERK1/2 (NPM1 vs. NPM1+LPS-RS, 3-fold-decrease,
P<0.05; NPM1 vs. NPM1+TAK242, 16-fold-decrease, P<0.01),
which suggests that NPM1 signaling through TLR4/MD-2 activated the
ERK1/2 signaling pathway.
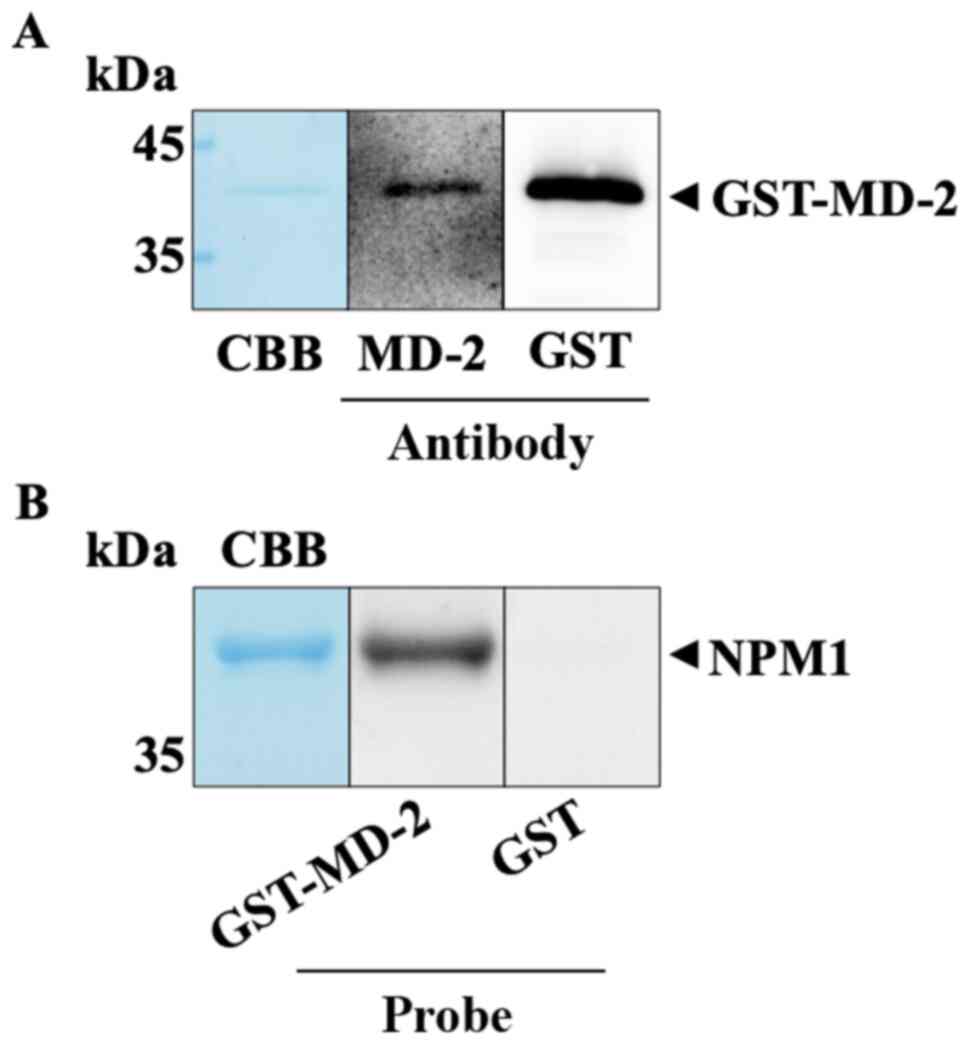
NPM1 binds MD-2
Next, far-western blotting was used to determine
whether NPM1 directly binds MD-2. GST-MD-2 was purified and
detected as a single band in CBB-stained gels (Fig. 3A, left), corresponding to the
specific band detected by the anti-MD-2 antibody (Fig. 3A, middle) and the anti-GST antibody
(Fig. 3A, right) in western blotting
experiments. NPM1 was subjected to SDS-PAGE, and then transferred
to a nitrocellulose membrane. The membrane was incubated with
GST-MD-2 or GST for 16 h at 4˚C, and then with the anti-GST
antibody for 1 h at room temperature. GST-MD-2 bound NPM1 on the
membrane at the respective position in the CBB-stained gels, but
GST was not detectable (Fig. 3B).
These results showed that NPM1 bound GST-MD-2, but not GST, which
suggests that NPM1 signaling is mediated via TLR4/MD-2.
Discussion
In the present study, PMA-differentiated THP-1 cells
were used to show that TLR4/MD-2 serves as a receptor for the
extracellular DAMP, NPM1. The interaction between NPM1 and
TLR4/MD-2 was detected using a SEAP reporter gene assay. The MD-2
antagonist, LPS-RS, and the TLR4 signaling inhibitor, TAK-242,
significantly inhibited NPM1-induced TNF-α production and ERK1/2
activation. Furthermore, far-western blotting analysis revealed
that NPM1 directly bound MD-2, which further indicates that NPM1
signaling was mediated through the activation of TLR4/MD-2
signaling.
Host cells infected with Gram-negative bacteria are
exposed to LPS, which strongly induces the activation of TLR4/MD-2
signaling in specific target cells such as macrophages and
monocytes (21,22). TLR4/MD-2 signaling recruits MyD88,
which leads to activation of the MAPKs and NF-κB signaling
pathways. Furthermore, TLR4 ligands include cellular proteins that
act as DAMPs, such as histones H3 and H4 as well as HMGB1 (10-12),
which suggests that TLR4 signaling may serve as a therapeutic
target for treating inflammatory diseases, such as sepsis and RA.
However, the full spectrum of TLR4 ligands remains to be
identified.
In our previous study, it was shown that NPM1 may
act as a DAMP (5). NPM1 induces the
production of the proinflammatory cytokines, TNF-α, IL-6 and IL-8,
which in turn activates MAPKs, such as ERK1/2, p38 MAPK and JNK1/2.
ERK1/2 activation contributes to the induction of TNF-α (5). Moreover, NPM1 is detected in the serum
in a rat model of sepsis (CLP), which suggests that NPM1 may act as
a DAMP (5). Additionally, it was
shown that NPM1, but not heat-inactivated NPM1, induced TNF-α
production, which suggests that TNF-α production was induced by
NPM1, but not by other bacterial components.
In the present study, NPM1 signaling, indicated by
TNF-α production, was mediated via the TLR4/MD-2 signaling pathway.
Firstly, TNF-α production stimulated by NPM1 was significantly
inhibited by the MD-2 antagonist, LPS-RS. LPS-RS, which is a
penta-acylated LPS, was identified as a potent antagonist of
LPS-induced toxicity (14). LPS-RS
is a competitive inhibitor of LPS via direct binding to MD-2
(14-17).
More recently, it has been shown that the dimerization of TLR4/MD-2
is essential for activation of TLR4 signaling. Indeed, the
dimerization ratio of TLR4/MD-2 increased by 48% in LPS-stimulated
cells (23). However, treatment with
LPS-RS completely inhibited this dimerization (23), thereby inhibiting TLR4/MD-2
signaling. Additionally, in the present study, TNF-α production by
NPM1 stimulation was significantly inhibited by TAK-242. The
cyclohexane derivative, TAK-242, selectively inhibits TLR4
signaling (15) through binding to
the TIR domain of TLR4 via Cys747(16). Binding of TAK-242 to the TLR4
intracellular domain disrupts the interaction of TLR4 with its
adaptor molecules TIR domain-containing adaptor protein and TIR
domain-containing adaptor including interferon-β-related adaptor
molecule (17). Furthermore,
far-western blotting demonstrated that NPM1 directly bound MD-2,
and thus NPM1 may induce TLR4/MD-2 dimerization. Finally, NPM1 has
been shown to activate MAPKs, including ERK1/2, JNK and p38 MAPK
(5). However, TNF-α production was
only mediated via ERK1/2, but not the other kinases (5). The present study examined whether
TAK-242 and LPS-RS inhibited NPM1-induced ERK1/2 activation. It was
shown that both inhibitors significantly inhibited NPM1-stimulated
ERK1/2 activation. Consistently, ERK1/2 was not activated in
LPS-treated THP-1 cells transfected with TLR4 small
interfering RNA (24). Furthermore,
TNF-α production has been shown to be mediated via ERK1/2
activation by TLR4/MD-2 in vitro and in vivo
(25,26). Taken together, the results indicate
that TNF-α production by NPM1 stimulation may be mediated via the
TLR4/MD-2/ERK1/2 signal transduction pathway.
NPM1 belongs to a chaperone family, which comprises
multiple major functional members (NPM1, NPM2 and NPM3) in the
intracellular space (27). Residues
in the N-terminal domain (Met9 to Asp122) are highly conserved and
essential for its oligomerization and interactions with other
proteins (8). Furthermore, NPM1,
which exists as a pentamer via the N-terminal domain, interacts
with other pentamers, and two NPM1 pentamers interact in a
head-to-head manner to form a decamer in the nucleus. The decamer
is modulated by numerous post-translational modifications,
especially phosphorylation (28). In
the extracellular space, the pentamer structure of NPM1 may be
essential because heat-inactivated NPM1 did not induce TNF-α
production. However, the NPM1 decamer did not affect the
interaction between NPM1 and MD-2 in the far-western blotting.
These findings suggest that the interaction between NPM1 and TLR4
may be mediated via the decamer form of NPM1, but not the
interaction between NPM1 and MD-2. This discrepancy will be
investigated in future studies.
To the best of our knowledge, the present study is
the first report to demonstrate that the NPM1 receptor is
TLR4/MD-2. NPM1 released from damaged or activated cells
potentially exacerbate the effects of inflammatory diseases.
Furthermore, NPM1 may contribute to the accumulation of
inflammatory cells in TLR4-related inflammatory diseases, such as
arterial thrombosis, RA, atherosclerosis and type II diabetes
mellitus as well as sepsis (29-32).
These findings may contribute to efforts to develop novel and more
effective treatments for inflammatory diseases by targeting TLR4
and components of its signaling pathway.
Acknowledgements
We would like to thank Miss Kazumi Tamura, Mrs.
Kazumi Sugiyama, and Mrs. Misako Tsujimoto for their excellent
technical assistance.
Funding
This study was supported by a Grant-in-aid from
KAKENHI (grant no. 18H02906).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KN, HU, ST, KK, NM, TO, EOA, KM, YoM, HI, IM, and
KIK conceived and designed the study, and were responsible for the
interpretation of the results and writing the manuscript. KN, HU,
YI, RCS, YuM, IK, and ST analyzed and interpreted the data and
helped prepare the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ito T, Totoki T, Yokoyama Y, Yasuda T,
Furubeppu H, Yamada S, Maruyama I and Kakihana Y: Serum histone H3
levels and platelet counts are potential markers for coagulopathy
with high risk of death in septic patients: A single-center
observational study. J Intensive Care. 7(63)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Roh JS and Sohn DH: Damage-associated
molecular patterns in inflammatory diseases. Immune Netw.
18(e27)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kikuchi K, Kawahara K, Tancharoen S,
Matsuda F, Morimoto Y, Ito T, Biswas KK, Takenouchi K, Miura N,
Oyama Y, et al: The free radical scavenger edaravone rescues rats
from cerebral infarction by attenuating the release of
high-mobility group box-1 in neuronal cells. J Pharmacol Exp Ther.
329:865–874. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Morimoto Y, Kawahara KI, Tancharoen S,
Kikuchi K, Matsuyama T, Hashiguchi T, Izumi Y and Maruyama I: Tumor
necrosis factor-alpha stimulates gingival epithelial cells to
release high mobility-group box 1. J Periodontal Res. 43:76–83.
2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nawa Y, Kawahara K, Tancharoen S, Meng X,
Sameshima H, Ito T, Masuda Y, Imaizumi H, Hashiguchi T and Maruyama
I: Nucleophosmin may act as an alarmin: Implications for severe
sepsis. J Leukoc Biol. 86:645–653. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xu J, Zhang X, Pelayo R, Monestier M,
Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F and Esmon CT:
Extracellular histones are major mediators of death in sepsis. Nat
Med. 15:1318–1321. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Box JK, Paquet N, Adams MN, Boucher D,
Bolderson E, O'Byrne KJ and Richard DJ: Nucleophosmin: From
structure and function to disease development. BMC Mol Biol.
17(19)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Borer RA, Lehner CF, Eppenberger HM and
Nigg EA: Major nucleolar proteins shuttle between nucleus and
cytoplasm. Cell. 56:379–390. 1989.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang H, Wang H, Ju Z, Ragab AA, Lundbäck
P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, et al: MD-2 is
required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med.
212:5–14. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xu J, Zhang X, Monestier M, Esmon NL and
Esmon CT: Extracellular histones are mediators of death through
TLR2 and TLR4 in mouse fatal liver injury. J Immunol.
187:2626–2631. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
He M, Bianchi ME, Coleman TR, Tracey KJ
and Al-Abed Y: Exploring the biological functional mechanism of the
HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol Med.
24(21)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kuzmich NN, Sivak KV, Chubarev VN, Porozov
YB, Savateeva-Lyubimova TN and Peri F: TLR4 signaling pathway
modulators as potential therapeutics in inflammation and sepsis.
Vaccines (Basel). 5(34)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Coats SR, Pham TT, Bainbridge BW, Reife RA
and Darveau RP: MD-2 mediates the ability of tetra-acylated and
penta-acylated lipopolysaccharides to antagonize Escherichia
coli lipopolysaccharide at the TLR4 signaling complex. J
Immunol. 175:4490–4498. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Takashima K, Matsunaga N, Yoshimatsu M,
Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y and Ii M:
Analysis of binding site for the novel small-molecule TLR4 signal
transduction inhibitor TAK-242 and its therapeutic effect on mouse
sepsis model. Br J Pharmacol. 157:1250–1262. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Matsunaga N, Tsuchimori N, Matsumoto T and
Ii M: TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like
receptor (TLR) 4 signaling, binds selectively to TLR4 and
interferes with interactions between TLR4 and its adaptor
molecules. Mol Pharmacol. 79:34–41. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Visintin A, Halmen KA, Latz E, Monks BG
and Golenbock DT: Pharmacological inhibition of endotoxin responses
is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol.
175:6465–6472. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Park EK, Jung HS, Yang HI, Yoo MC, Kim C
and Kim KS: Optimized THP-1 differentiation is required for the
detection of responses to weak stimuli. Inflamm Res. 56:45–50.
2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fujita H, Yagishita N, Aratani S,
Saito-Fujita T, Morota S, Yamano Y, Hansson MJ, Inazu M, Kokuba H,
Sudo K, et al: The E3 ligase synoviolin controls body weight and
mitochondrial biogenesis through negative regulation of PGC-1β.
EMBO J. 34:1042–1055. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Walsh BW, Lenhart JS, Schroeder JW and
Simmons LA: Far western blotting as a rapid and efficient method
for detecting interactions between DNA replication and DNA repair
proteins. Methods Mol Biol. 922:161–168. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Akashi S, Shimazu R, Ogata H, Nagai Y,
Takeda K, Kimoto M and Miyake K: Cutting edge: Cell surface
expression and lipopolysaccharide signaling via the toll-like
receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol.
164:3471–3475. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Horvatinovich JM, Grogan EW, Norris M,
Steinkasserer A, Lemos H, Mellor AL, Tcherepanova IY, Nicolette CA
and DeBenedette MA: Soluble CD83 inhibits T cell activation by
binding to the TLR4/MD-2 complex on CD14+ monocytes. J
Immunol. 198:2286–2301. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Krüger CL, Zeuner MT, Cottrell GS, Widera
D and Heilemann M: Quantitative single-molecule imaging of TLR4
reveals ligand-specific receptor dimerization. Sci Signal.
10(eaan1308)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Singh A, Singh V, Tiwari RL, Chandra T,
Kumar A, Dikshit M and Barthwal MK: The IRAK-ERK-p67phox-Nox-2 axis
mediates TLR4, 2-induced ROS production for IL-1β transcription and
processing in monocytes. Cell Mol Immunol. 13:745–763.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Smith JA, Stallons LJ, Collier JB, Chavin
KD and Schnellmann RG: Suppression of mitochondrial biogenesis
through toll-like receptor 4-dependent mitogen-activated protein
kinase kinase/extracellular signal-regulated kinase signaling in
endotoxin-induced acute kidney injury. J Pharmacol Exp Ther.
352:346–357. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Luan H, Zhang Q, Wang L, Wang C, Zhang M,
Xu X, Zhou H, Li X, Xu Q, He F, et al: OM85-BV induced the
productions of IL-1β, IL-6, and TNF-α via TLR4- and TLR2-mediated
ERK1/2/NF-κB pathway in RAW264.7 cells. J Interferon Cytokine Res.
34:526–536. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cela I, Di Matteo A and Federici L:
Nucleophosmin in its interaction with ligands. Int J Mol Sci.
21(4885)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mitrea DM, Grace CR, Buljan M, Yun MK,
Pytel NJ, Satumba J, Nourse A, Park CG, Madan Babu M, White SW, et
al: Structural polymorphism in the N-terminal oligomerization
domain of NPM1. Proc Natl Acad Sci USA. 111:4466–4471.
2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen X, Tao T, Wang H, Zhao H, Lu L and Wu
F: Arterial thrombosis is accompanied by elevated mitogen-activated
protein kinase (MAPK) and Cyclooxygenase-2 (COX-2) expression via
Toll-like receptor 4 (TLR4) activation by S100A8/A9. Med Sci Monit.
24:7673–7681. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Watanabe T, Takahashi N, Hirabara S,
Ishiguro N and Kojima T: Hyaluronan inhibits TLR4-dependent RANKL
expression in human rheumatoid arthritis synovial fibroblasts. PLoS
One. 11(e0153142)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Blich M, Golan A, Arvatz G, Sebbag A,
Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S,
Eitan A, et al: Macrophage activation by heparanase is mediated by
TLR-2 and TLR-4 and associates with plaque progression.
Arterioscler Thromb Vasc Biol. 33:e56–e65. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lu Z, Zhang X, Li Y, Lopes-Virella MF and
Huang Y: TLR4 antagonist attenuates atherogenesis in LDL
receptor-deficient mice with diet-induced type 2 diabetes.
Immunobiology. 220:1246–1254. 2015.PubMed/NCBI View Article : Google Scholar
|