Introduction
Cocaine, a potent CNS-stimulant, is abused
predominantly in Western countries. Current estimates indicate that
>5.9 million Americans used this drug in 2018(1), and >18 million individuals have used
it worldwide (2). Cocaine entry in
the brain initiates a variety of responses ranging from toxicity to
altered signal transduction in different CNS cell-types (3). However, despite the fact that the brain
is composed of neurons, astrocytes, microglia and oligodendrocytes,
the majority of in vitro or in vivo studies with
cocaine have been focused on neurons (4,5), partly
due to the interactions of cocaine at the signal transduction
level.
Astrocytes are one of the most abundant cell-types
in the CNS, functioning in neuronal survival and maintenance of
fundamental patterns of circuitry (6). It is well established that astrocytes
mediate synaptic cross-talk (7),
suggesting that astrocytes may be just as important as neurons in
research on cocaine abuse. Owing to their abundance in the majority
of regions of the brain (8),
astrocytes may be the first type of cells to experience the toxic
effects of cocaine through decreased mitochondrial membrane
potential, resulting in a hypoxia-like state in cells, with regard
to being unable to reduce oxygen to water. The hypoxic condition in
turn induces the expression of HIF-1α and VEGF in cells and
triggers inflammation.
NO is product of inflammatory responses, generated
by inducible-nitric oxide synthase (iNOS) in astrocytes (9) under hypoxic conditions (10). In vivo, NO is produced from
L-arginine by the action of the constitutively active form of NOS
due to activation of N-methyl-D-aspartate (NMDA) receptors on
glutamatergic neurons. Conversely, iNOS in astrocytes (9) is also involved in NO production from
L-arginine through the stimulation of NMDA receptors (11). A previous in vitro study
showed that cocaine did not stimulate NO production in resting
(unstimulated) cells (12). This
observation indicated the need of a certain physiological stimulus
for NO release. Astrocytes exposed to external insults/challenges
function as immunocompetent cells (13) and release pro or anti-inflammatory
signals. Since an unhealthy state of the physical body (for
example, bacterial infection) results in altered physiology, it is
possible that astrocytes may initiate an immune response by
releasing pro or anti-inflammatory molecules (14). Under such a state, it is not clear
whether cocaine would potentiate the rate at which NO is produced
in the CNS. Excess NO release has been shown to be a contributing
factor to several CNS disorders, such as Parkinson's disease
(15), schizophrenia or Alzheimer's
disease. To date, no studies have determined whether cocaine can
potentiate inflammation in astrocytes, to the best of our
knowledge.
In the present study, the role of cocaine on the
production of different inflammatory products, such as
O2- free radicals,
H2O2, NO2-, (a stable
product of NO), HIF-1α and VEGF was assessed in rat C6
astroglia-like cells stimulated with LPS (endotoxin) and IFNγ
(pro-inflammatory cytokine). These cells are astrocytic in origin
and exhibit a high degree of similarity with human astrocytes in
terms of gene expression (16), and
have been employed frequently in drug abuse studies (12,17-20).
Materials and methods
Materials
DMEM, heat inactivated FBS, Hank's balanced salt
solution (HBSS) and PBS were purchased from Bio-Rad Laboratories,
Inc. Penicillin/streptomycin sulfate, amphotericin B and
L-glutamine were purchased from Mediatech, Inc. Cocaine
hydrochloride (Ecgonine methyl ester benzoate; molecular weight,
339.8), trypan blue, sulfanilamide, LPS from Escherichia
coli serotype 0111:B4, N-(1-naphthyl)-ethylenediamine (NED),
phosphoric acid, and EDTA were supplied by Sigma-Aldrich; Merck
KGaA. IFNγ protein was obtained from Novus Biologicals Ltd. All
other routinely used agents were analytical grade.
Cell culture
C6 astrocyte-like cell line (CCL-107) was
purchased from American Type Culture Collection and maintained as
an adherent monolayer culture in complete DMEM in phenol red
supplemented with 2 mM L-glutamine, 10% (v/v) FBS, 100 U/ml
penicillin, 100 μg/ml streptomycin sulfate and 0.25 µg/ml
amphotericin B. Cells were grown in a humidified incubator at 37˚C
with 5% CO2, and sub-cultured twice a week. For
cytotoxic studies, the culture was harvested by treating with 0.05%
EDTA in PBS for ≤2 min, resulting in a single cell suspension. Cell
count was assessed using 0.4% trypan blue dye exclusion assay (27˚C
for 1 min) using a hemocytometer under a light microscope
(magnification, x20).
Viability assay
Cells (5x104/well) in 96-well plates were
treated with cocaine (1-4 mM) for 24 h. Cocaine stock, working
stocks and treatments were prepared as described previously
(17). Cell viability was evaluated
using a crystal violet dye uptake assay as described previously
(17). At the end of treatment, 100
µl 0.25% glutaraldehyde was added per well and incubated for 30
min. After washing and air drying of plates, the dye was extracted
with 100 µl 50 mM sodium phosphate mono basic solution, containing
50% ethyl alcohol. The plates were gently vortexed and the optical
density (OD) measurements of incorporated dye in viable cells were
measured at 540 nm using a microplate spectrophotometer (Bio-Tek
Instruments Inc.).
Superoxide assay
Cells (5x104/well) in 96-well plates in
phenol red free media were treated with LPS (0.2 µg/ml), IFNγ (6
µg/ml) and cocaine (1-4 mM) for 24 h. Production of superoxide
radicals from cells was detected using a cytochrome c
reduction assay as described previously (21). Briefly, at the end of the 24 h
treatment, the supernatant from samples was added to an equivalent
volume of cytochrome c from horse heart (Sigma-Aldrich;
Merck KGaA) prepared in PBS (final working concentration: 160 µM).
Then the samples were incubated for 35 min at 37˚C with 5%
CO2. OD measurements of samples were obtained at 550 nm
using a UV microplate spectrophotometer.
Hydrogen peroxide assay
Cells (5x104/well) in 96-well plates in
phenol red free media were treated as described above, and
H2O2 production was assessed using a
previously described method with some modifications (22). After 24 h of treatment, sample
supernatants were assessed for H2O2
production using a peroxidase linked continuous assay. The
chromogenic solution contained (final working concentration) 1 mM
vanillic acid, 500 µM 4-aminoantipyrine and horseradish peroxidase
(4 U/ml) in PBS with 2 mM HEPES (pH 7.4). The chromogenic solution
was added to each sample and incubated for 10 min at 37˚C. Samples
were analyzed at 490 nm in a UV micro plate spectrophotometer
(Bio-Tek Instruments Inc.). Controls and blanks were measured
simultaneously, and subtracted from the final value to eliminate
interference.
NO/NO2-
determination
In order to determine the optimum concentrations of
IFNγ (2.6, 3.3, 4.0, 4.6, 5.3, 6.0, 6.6 7.3, 8.0 and 8.6 µg/ml) or
LPS (2.6, 3.3, 4.0, 4.6, 5.3, 6.0, 6.6 7.3, 8.0 and 8.6 µg/ml), a
dose-response study was initially performed in 96-well plates.
Then, the role of cocaine on NO generation was studied by treating
the cells in phenol red free media as described above. At the end
of treatments, Griess reagent (mixture of an equal volume of 1%
sulfanilamide in 0.5 N HCl and 0.1% NED in deionized water) was
added directly to the cells under reduced lighting at room
temperature. The plates were read at 540 nm on a UV microplate
spectrophotometer. Controls and blanks were measured
simultaneously, and subtracted from the final value to eliminate
interference. A standard curve was generated from a range of
dilutions of sodium nitrite (1-100 µM) prepared in the plating
medium.
HIF-1α ELISA
For quantification of HIF-1α release in cell
lysates, the cells (5x104/well) were seeded in 96-well
plates with DMEM. Supernatants from resting (unstimulated) and
stimulated (with LPS and IFNγ) cells after 24 h cocaine exposure at
different concentrations (1-4 mM) were discarded, and ice cold PBS
was immediately added to each well, decanted and 100 µl cell lysis
buffer (Abcam) supplemented with 1:500 µl protease inhibitor (cat.
no. ab65621; Abcam) and N-methoxyoxoacetyl-glycine methyl ester
(Sigma-Aldrich; Merck KGaA; cat. no. D3695, lot#:064M4728V) was
added to each well. After three freeze/thaw cycles at -80˚C, the
content of each well was centrifuged at 100 x g for 5 min at 4˚C.
Then, 90 µl supernatant (centrifuged vial) was added to each ELISA
well. ELISA (kit sensitivity, 0.78 pg/ml) was performed using a
HIF-1α ELISA kit (CUSABIO; cat. no. E08540r) according to the
manufacturer's protocol. Briefly, 90 µl supernatant from samples
and standards were added to 96-well plates pre-coated with the
capture antibody. After incubation for 2 h at 37˚C, the supernatant
from each well was decanted and 100 µl of the prepared biotinylated
primary antibody mixture was added. After 1 h of incubation at
37˚C, all wells were washed 3 times with 300 µl 1X wash buffer
provided in the kit. The wash buffer was discarded and 100 µl
prepared secondary antibody, HRP-avidin, was added to each well and
incubated for 1 h at 37˚C. Subsequently, all wells were washed 5
times with 300 µl 1X wash buffer provided in the kit. After removal
of the washing buffer, 90 µl of the color developing agent was
added to each well and incubated at 37˚C for 25 min. Finally, 50 µl
stop solution was added to each well and the absorbance was read at
450 nm using a UV microplate reader.
VEGF ELISA
For quantification of VEGF release in cell lysates,
the cells (5x104/well) were seeded in 96-well plates
with DMEM. After discarding the supernatant, the cell lysate was
prepared as described for the HIF-1α ELISA. ELISA (kit sensitivity,
20 pg/ml) was performed using a VEGF ELISA kit (Boster Biological
Technology; cat. no. EK0540) according to the manufacturer's
protocol. Briefly, 100 µl of the samples and standards were added
to 96-well plates pre-coated with the capture antibody. After
incubation for 90 min at 37˚C, the supernatant from each well was
decanted and 100 µl of the prepared biotinylated primary antibody
mixture was added. After 1 h of incubation at 37˚C, all wells were
washed 3 times with 300 µl 0.01 M PBS. The wash buffer was
discarded and 100 µl of the prepared secondary antibody,
Avidin-Biotin- Peroxidase Complex, was added to each well and
incubated for 30 min at 37˚C. Subsequently, all wells were washed 5
times with 300 µl 0.01 M PBS. After removal of the washing buffer,
90 µl color developing agent was added to each well and incubated
at 37˚C for 25 min. Finally, 100 µl stop solution was added to each
well and the plate was read at 450 nm using an UV microplate
reader.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Differences between groups were compared using a one-way
ANOVA followed by a post-hoc Dunnett's tests. Data analysis was
performed using GraphPad Version 6 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
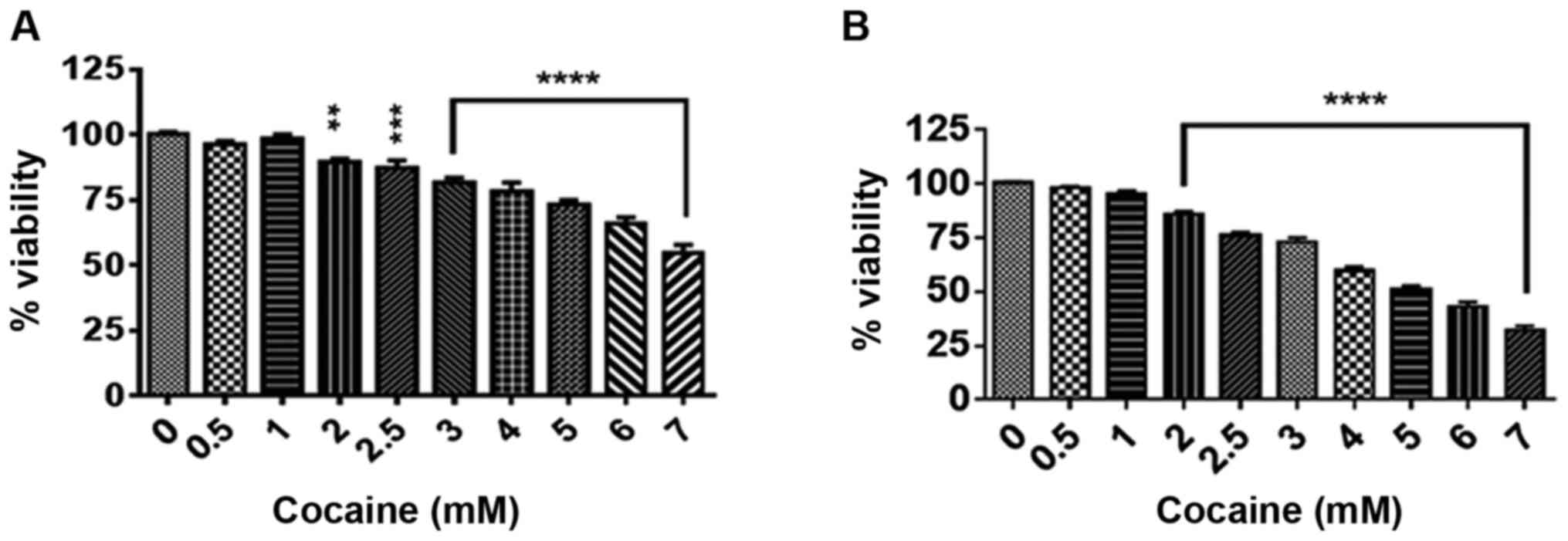
Cocaine decreases cell viability
Initially the effect of treatment with cocaine at
concentrations ranging from 0.5 to 7 mM for 24 and 48 h on cell
viability was assessed. The selection of cocaine concentrations was
based on earlier reports (20,23), and
the lag period and doubling time of these cells (17) were used as the criteria for treatment
durations of 24 and 48 h. Compared with the corresponding controls,
cocaine significantly decreased the cell viability at these time
points (Fig. 1A and B; P<0.01 or P<0.001). The
LC50, where 50% of cells were killed by cocaine, was
determined to be between 4.5 and 3.8 mM at 24 and 48 h,
respectively.
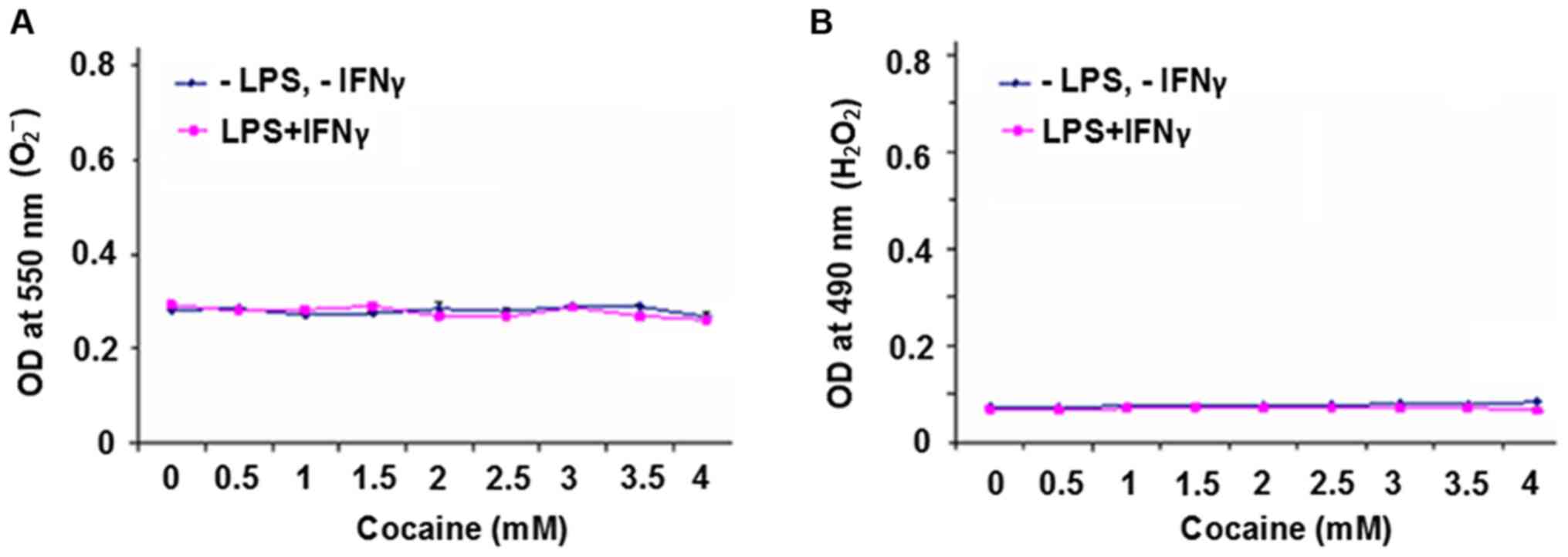
O2- free radical
and H2O2 production
Cells treated with cocaine for 48 h exhibited a
higher rate of death compared with 24 h of treatment (Fig. 1A and B); in order to prevent severe loss of
cells, 24 h of treatment was selected as the end point for
subsequent experiments. The effect of cocaine on the production of
O2- free radicals and
H2O2 in cells with or without LPS and IFNγ
stimulation was determined. It was found that there was no
difference between the unstimulated or stimulated groups
irrespective of cocaine treatment (P>0.05) for either of the
products (Fig. 2A and B).
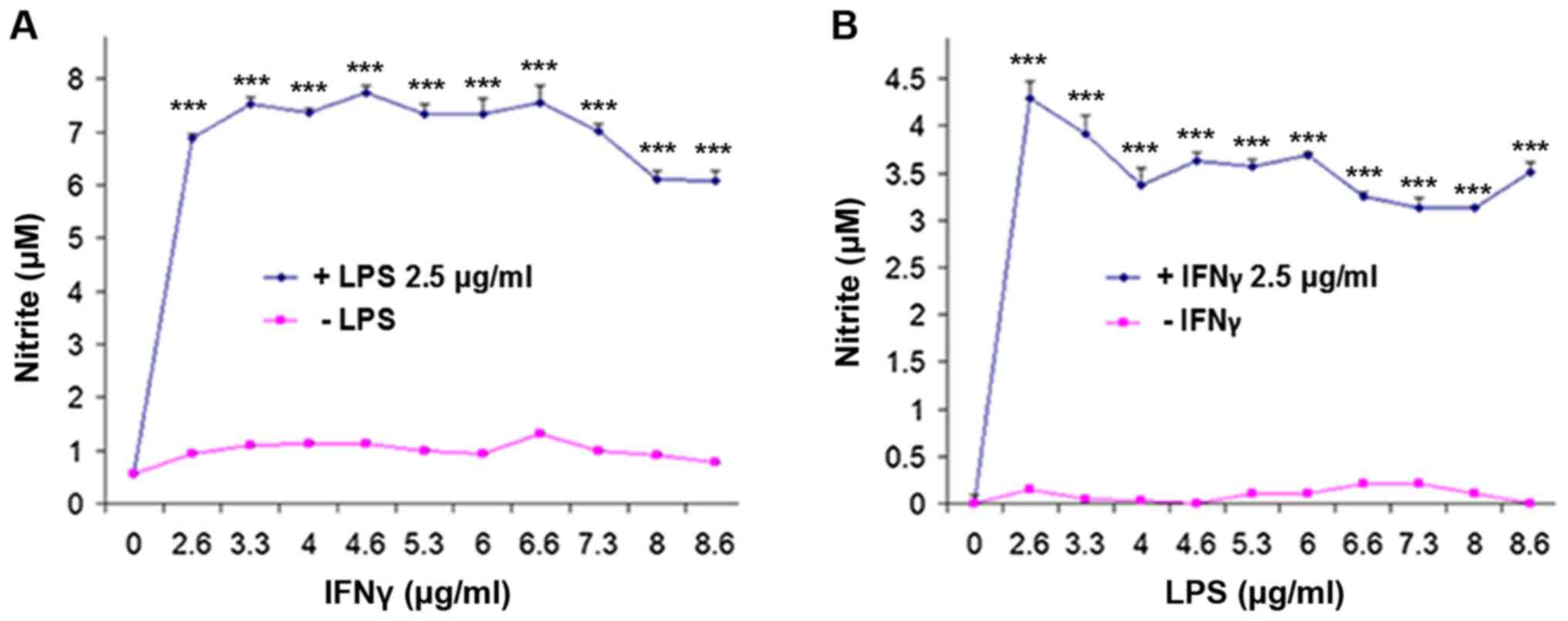
NO/NO2-
production
Next NO generation in cells was observed following
stimulation and treatment. To determine the optimum concentrations
of LPS and IFNγ, the cells were initially treated for 24 h with
increasing concentrations of IFNγ against a fixed concentration
(2.5 µg/ml) of LPS (Fig. 3A).
Similarly, a dose-response experiment was performed for LPS was
performed for 24 h against a fixed concentration (2.5 µg/ml) of
IFNγ (Fig. 3B). It was found that
0.2 µg/ml LPS and 6 µg/ml IFNγ were ideal to stimulate
NO/NO2- production without compromising the
cell viability (data not shown), consistent with a previous study
(24). Based on this, the cells were
stimulated with LPS and IFNγ at these doses, and treated with
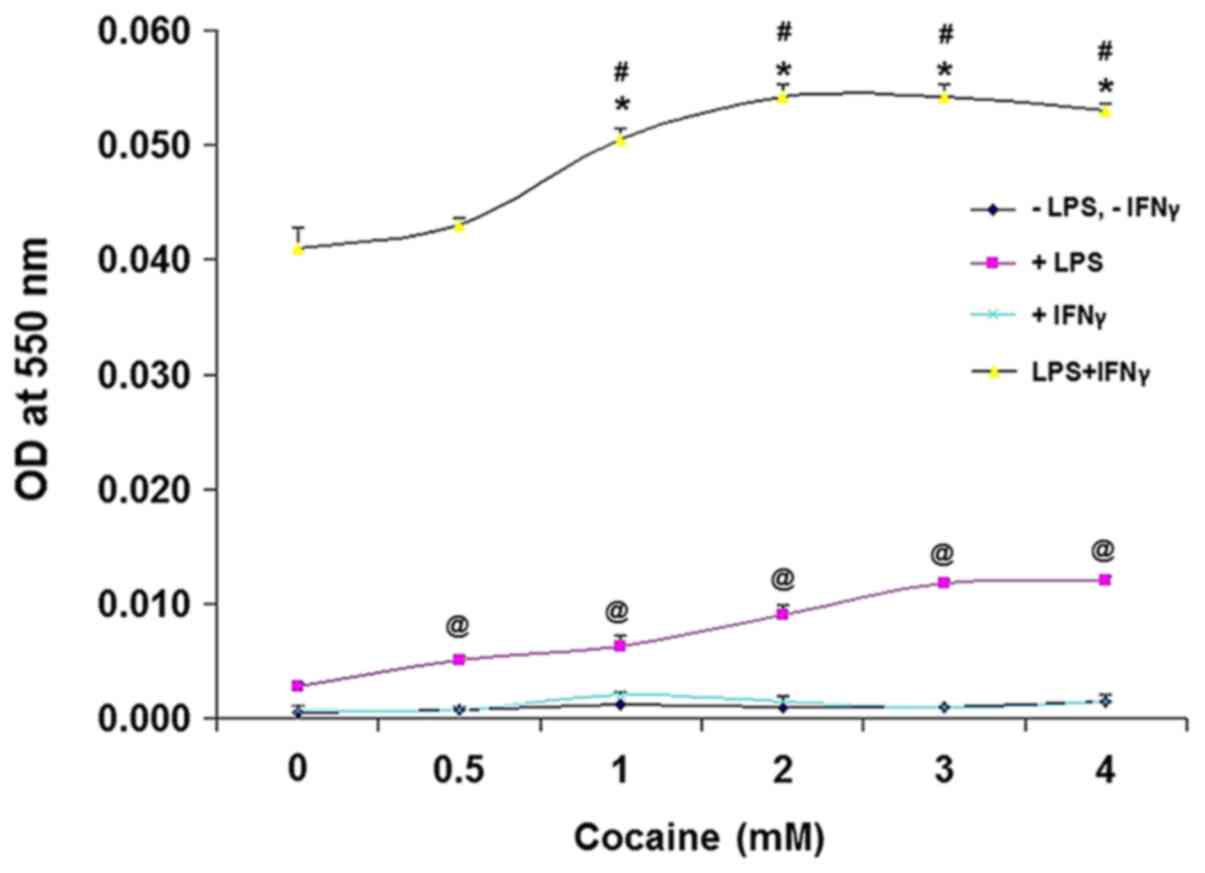
cocaine for 24 h. Lack of LPS and IFNγ stimulation did not result
in altered NO/NO2- production in the cocaine
treated cells (Fig. 4); however,
when stimulated with LPS alone, cocaine at different concentrations
increased NO/NO2- production significantly in
a dose-dependent manner compared with the unstimulated
cocaine-treated cells (P<0.01), corroborating a previous report
on LPS-induced NO/NO2- production (25). Conversely, IFNγ-stimulated cells did
not produce NO/NO2- following cocaine
treatment. However, LPS and IFNγ-stimulated cells treated with
cocaine showed a significant increase in
NO/NO2- production (40-50x; absorbance, 0.04
to 0.05, respectively; Fig. 4)
compared with the unstimulated cells (absorbance, 0.001; Fig. 4; P<0.001).
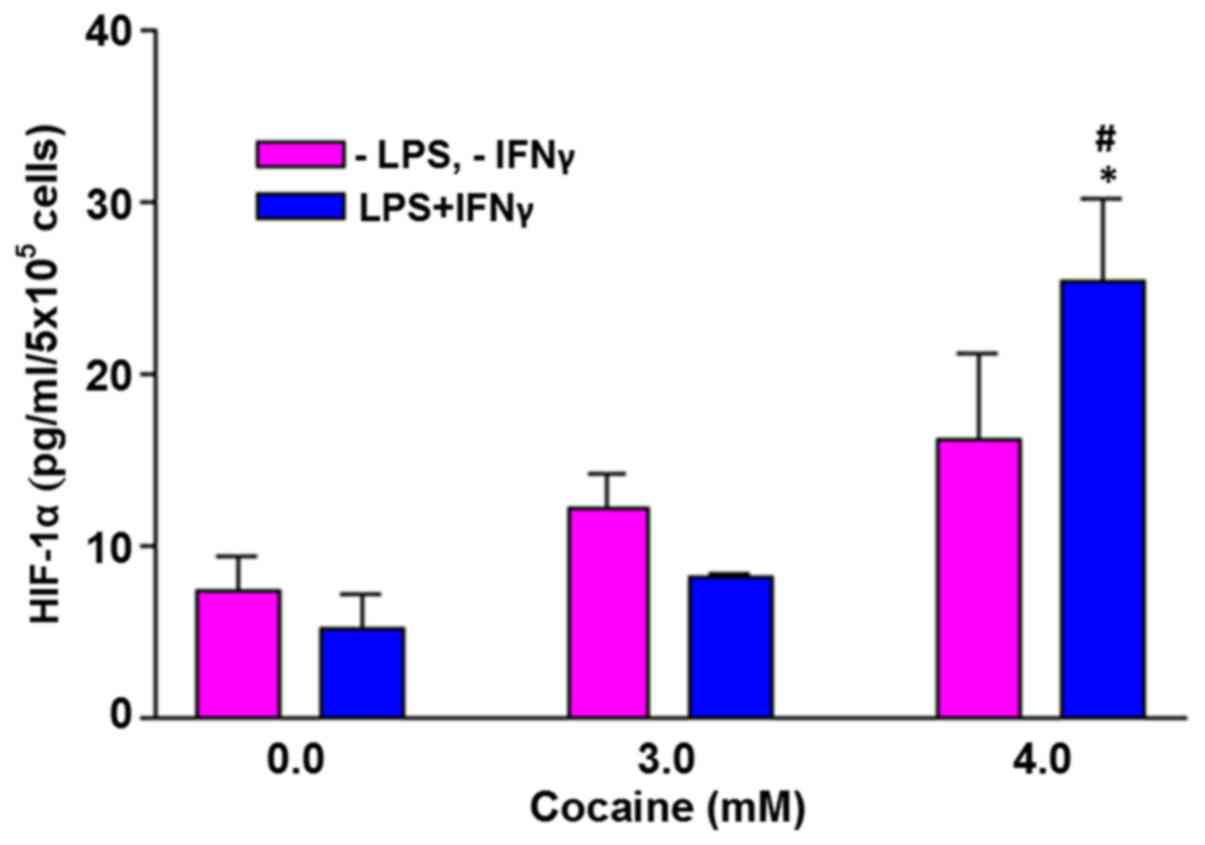
HIF-1α levels are increased by
treatment with cocaine in the stimulated cells
In the absence of external challenge to the cells,
there was no significant increase in HIF-1α with cocaine treatment
at 3 and 4 mM (Fig. 5). However,
when the cells were challenged with LPS and IFNγ, the HIF-1α levels
in cells treated with 4 mM cocaine increased significantly by
5-fold (P<0.05). The average increase was 25.3±4.9 pg/ml of the
control (5.1±2.1 pg/ml).
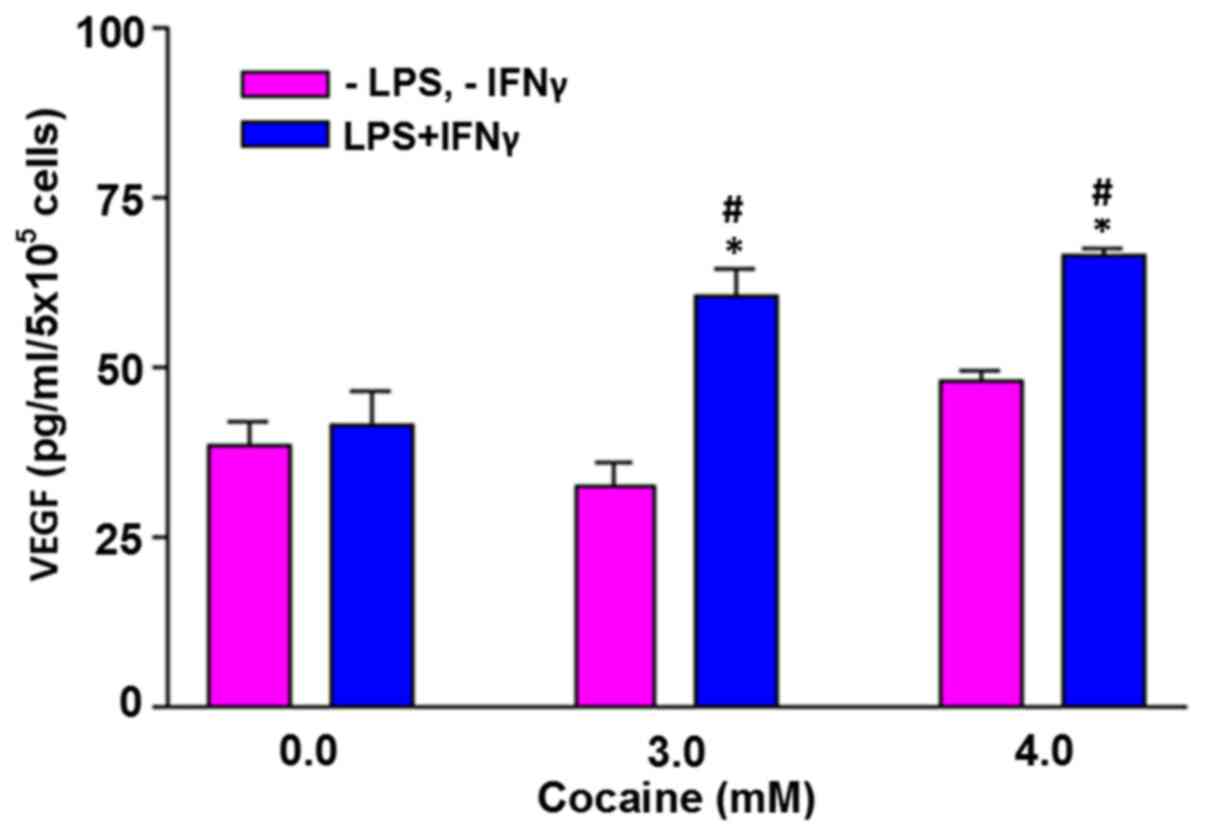
VEGF levels are increased by treatment
with cocaine in the stimulated cells
In the absence of external challenge, cells treated
with cocaine at 3 and 4 mM did not increase VEGF levels (Fig. 6). However, when the cells were
challenged with LPS and IFNγ, the VEGF levels in the cells treated
with 3 and 4 mM cocaine increased significantly (P<0.05).
Compared with the control (41.8±4.5), the average increase was
60.5±3.9 and 66.4±1.1 pg/ml, respectively at 3 and 4 mM
cocaine.
Discussion
Consistent with an earlier report (17), cocaine resulted in a dose and
time-dependent cell death. Studies have shown that cocaine-induced
cellular dysfunction was associated with increased production of
reactive oxygen species (20,26) and
H2O2 (26,27).
However, in the present study, cocaine treatment did not increase
the production of these oxidative species, which may have been due
to either a difference in the cell model used, or a longer period
of exposure compared with acute treatment (20).
The ubiquitous biological molecule NO is a small
molecule, and a volatile gas with several molecular targets in the
body (28). It has both beneficial
and harmful effects; for example, at physiological concentrations,
it is associated with neurotransmission (29), cognitive function and synaptic
plasticity. However, excess production is associated with
inflammation with detrimental consequences related to neurological
disorders (30,31). Thus, over production of NO represents
a diseased-state in the body.
In the present study, a diseased-state was simulated
by challenging the astroglia-like cells with endotoxin (LPS), which
is commonly found on the cell membrane of gram negative bacteria
(32), and a pro-inflammatory
cytokine (IFNγ). Using this challenge as a means of activating
astrocytes, whether cocaine potentiated the inflammatory response
was assessed. Increased NO/NO2- production
was observed in LPS and IFNγ stimulated cells in the absence of
cocaine, indicating a synergistic effect of LPS and IFNγ, an
observation consistent with an earlier study in the same cell line
(33). Additionally, the further
increase in the NO/NO2- levels following
cocaine treatment suggested that cocaine potentiates
NO/NO2- production under diseased-states.
Under normoxic conditions, certain enzymes, such as
prolyl-hydroxylases domain (PhD) degrade HIF-1α through the
proteasomal pathway. This way, the intracellular concentrations of
HIF-1α under normoxic conditions are maintained at a low level.
However, under a hypoxic environment, the PhD enzymes are
inhibited, leading to accumulation of stable HIF-1α in cells.
Similarly, a high concentration of NO (>1 µM) also stabilizes
HIF-1α under normoxic conditions (34). The presence of stable HIF-1α in the
cytoplasm in turn activates several genes required for adaptation
under conditions of low oxygen availability (35). One of the more relevant genes
targeted by stable HIF-1α is VEGF (36,37). In
the present study, detection of HIF-1α and VEGF following cocaine
treatment suggested the generation of the hypoxic environment in
cells was due to dysfunctional mitochondria owing to a loss of
membrane potential (17). The
presence of iNOS in astrocytes (9),
and production of NO by iNOS under hypoxic conditions (10) triggered HIF-1α and VEGF production in
cells (Figs. 5 and 6).
The physiological relevance of the results would
have been greater if primary astrocytes were used in the present
study; but due to their limited growth potential, finite life span
and lack of cell homogeneity between different primary cultures,
C6 astroglia-like cells were instead used. These cells
exhibit a high degree of similarity with human astrocytes (16); thus, the results of the present study
may also have in vivo significance. Several studies have
shown the link between inflammation and drug abuse in the past; for
example, elevated levels of pro-inflammatory factors were observed
in cocaine-dependent individuals (38-41),
but no link was established with NO production. Whilst previous
reports in non-CNS cells showed cocaine-induced changes in NO
levels (42,43), no studies have reported on the effect
of cocaine on NO production in astrocytes under a diseased state,
to the best of our knowledge.
In conclusion, the present study showed that cocaine
potentiated the inflammatory response in LPS and IFNγ-stimulated
astroglia-like cells under a diseased-state. Since astrocytes
modulate the synaptic cross-talk in vivo (7), excess release of NO by activated
astrocytes with cocaine could result in dysfunction of neurons and
lead to the development or exacerbation of several CNS disorders
(44). The involvement of NMDA
receptors in excess NO production, their expression in astrocytes
(11) and glutamatergic neurons
highlights the possibility of using NMDA antagonists as a means of
regulating NO levels in the body; which may aid in delaying the
onset or progression of several CNS diseases.
Acknowledgements
We would like to thank Mrs. Veera L.D. Badisa
(Florida Agricultural and Mechanical University, School of the
Environment) for critically reading the manuscript and for
suggestions.
Funding
Funding: This study was supported by the National Institute on
Minority Health and Health Disparities of the National Institutes
of Health (grant no. U54MD007582).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MA, RBB, EM, CBG, and KFS conceived and designed the
study, and performed the experiments. RBB, EM and CBG interpreted
and analyzed the data and finalized the manuscript. All authors
read and approved the final manuscript. RBB, EM, and CBG confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Center for Behavioral Health Statistics
and Quality (CBHSQ): 2017 National Survey on Drug Use and Health:
detailed Tables. Substance Abuse and Mental Health Services
Administration, Rockville, MD, 2018.
|
|
2
|
United Nations Office on Drugs and Crime
(UNODC): World Drug Report 2018. United Nations publication, Sales
No. E.18.XI.9. UNODC, Vienna, 2018. https://www.unodc.org/wdr2018/.
|
|
3
|
Ritz MC, Lamb RJ, Goldberg SR and Kuhar
MJ: Cocaine receptors on dopamine transporters are related to
self-administration of cocaine. Science. 237:1219–1223.
1987.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Arencibia-Albite F, Vázquez-Torres R and
Jiménez-Rivera CA: Cocaine sensitization increases subthreshold
activity in dopamine neurons from the ventral tegmental area. J
Neurophysiol. 117:612–623. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Baimel C, McGarry LM and Carter AG: The
projection targets of medium spiny neurons govern cocaine-evoked
synaptic plasticity in the nucleus accumbens. Cell Rep.
28:2256–2263.e3. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Araque A: Astrocyte-neuron signaling in
the brain--implications for disease. Curr Opin Investig Drugs.
7:619–624. 2006.PubMed/NCBI
|
|
7
|
Roman C, Egert L and Di Benedetto B:
Astrocytic-neuronal crosstalk gets jammed: Alternative perspectives
on the onset of neuropsychiatric disorders. Eur J Neurosci: Jul 9,
2020 (Epub ahead of print).
|
|
8
|
Tsacopoulos M and Magistretti PJ:
Metabolic coupling between glia and neurons. J Neurosci.
16:877–885. 1996.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Raso GM, Meli R, Gualillo O, Pacilio M and
Di Carlo R: Prolactin induction of nitric oxide synthase in rat C6
glioma cells. J Neurochem. 73:2272–2277. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kawase M, Kinouchi H, Kato I, Akabane A,
Kondo T, Arai S, Fujimura M, Okamoto H and Yoshimoto T: Inducible
nitric oxide synthase following hypoxia in rat cultured glial
cells. Brain Res. 738:319–322. 1996.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lipton SA: NMDA receptors, glial cells,
and clinical medicine. Neuron. 50:9–11. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Badisa RB and Goodman CB: Effects of
chronic cocaine in rat C6 astroglial cells. Int J Mol Med.
30:687–692. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Colombo E and Farina C: Astrocytes: Key
regulators of neuroinflammation. Trends Immunol. 37:608–620.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tschaikowsky K, Hedwig-Geissing M, Schiele
A, Bremer F, Schywalsky M and Schüttler J: Coincidence of pro- and
anti-inflammatory responses in the early phase of severe sepsis:
Longitudinal study of mononuclear histocompatibility leukocyte
antigen-DR expression, procalcitonin, C-reactive protein, and
changes in T-cell subsets in septic and postoperative patients.
Crit Care Med. 30:1015–1023. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Schulz JB, Lindenau J, Seyfried J and
Dichgans J: Glutathione, oxidative stress and neurodegeneration.
Eur J Biochem. 267:4904–4911. 2000.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sibenaller ZA, Etame AB, Ali MM, Barua M,
Braun TA, Casavant TL and Ryken TC: Genetic characterization of
commonly used glioma cell lines in the rat animal model system.
Neurosurg Focus. 19(E1)2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Badisa RB, Darling-Reed SF and Goodman CB:
Cocaine induces alterations in mitochondrial membrane potential and
dual cell cycle arrest in rat c6 astroglioma cells. Neurochem Res.
35:288–297. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Badisa RB, Goodman CB and Fitch-Pye CA:
Attenuating effect of N-acetyl-L-cysteine against acute cocaine
toxicity in rat C6 astroglial cells. Int J Mol Med. 32:497–502.
2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Badisa RB, Fitch-Pye CA, Agharahimi M,
Palm DE, Latinwo LM and Goodman CB: Milk thistle seed extract
protects rat C6 astroglial cells from acute cocaine toxicity. Mol
Med Rep. 10:2287–2292. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Badisa RB, Kumar SS, Mazzio E, Haughbrook
RD, Allen JR, Davidson MW, Fitch-Pye CA and Goodman CB: N-acetyl
cysteine mitigates the acute effects of cocaine-induced toxicity in
astroglia-like cells. PLoS One. 10(e0114285)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pick E and Mizel D: Rapid microassays for
the measurement of superoxide and hydrogen peroxide production by
macrophages in culture using an automatic enzyme immunoassay
reader. J Immunol Methods. 46:211–226. 1981.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Holt A, Sharman DF, Baker GB and Palcic
MM: A continuous spectrophotometric assay for monoamine oxidase and
related enzymes in tissue homogenates. Anal Biochem. 244:384–392.
1997.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Badisa RB, Wi S, Jones Z, Mazzio E, Zhou
Y, Rosenberg JT, Latinwo LM, Grant SC and Goodman CB: Cellular and
molecular responses to acute cocaine treatment in neuronal-like N2a
cells: Potential mechanism for its resistance in cell death. Cell
Death Discov. 4(13)2018.PubMed/NCBI View Article : Google Scholar : Erratum in: Cell
Death Discov 5, 116 2019.
|
|
24
|
Zheng X, Liao Y, Wang J, Hu S, Rudramurthy
GR, Swamy MK, Rohit KC and Wang Y: The antineuroinflammatory effect
of simvastatin on lipopolysaccharide activated microglial cells.
Evid Based Complement Alternat Med. 2018(9691085)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Oliveira DM, Silva-Teixeira DN,
Araújo-Filho R and Goes AM: Antigenic stimulation is more efficient
than LPS in inducing nitric oxide production by human mononuclear
cells on the in vitro granuloma reaction in schistosomiasis. Braz J
Med Biol Res. 32:1437–1445. 1999.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhu W, Wang H, Wei J, Sartor GC, Bao MM,
Pierce CT, Wahlestedt CR, Dykxhoorn DM and Dong C: Cocaine exposure
increases blood pressure and aortic stiffness via the
miR-30c-5p-malic enzyme 1-reactive oxygen species pathway.
Hypertension. 71:752–760. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dalvi P, Wang K, Mermis J, Zeng R,
Sanderson M, Johnson S, Dai Y, Sharma G, Ladner AO and Dhillon NK:
HIV-1/cocaine induced oxidative stress disrupts tight junction
protein-1 in human pulmonary microvascular endothelial cells: Role
of Ras/ERK1/2 pathway. PLoS One. 9(e85246)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Moncada S, Palmer RMJ and Higgs EA: Nitric
oxide: Physiology, pathophysiology, and pharmacology. Pharmacol
Rev. 43:109–142. 1991.PubMed/NCBI
|
|
29
|
Schuman EM and Madison DV: Nitric oxide as
intercellular signal in long-term potentiation. Semin Neurosci.
5:207–215. 1994.
|
|
30
|
Good PF, Hsu A, Werner P, Perl DP and
Olanow CW: Protein nitration in Parkinson's disease. J Neuropathol
Exp Neurol. 57:338–342. 1998.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Guix FX, Wahle T, Vennekens K, Snellinx A,
Chávez-Gutiérrez L, Ill-Raga G, Ramos-Fernandez E, Guardia-Laguarta
C, Lleó A, Arimon M, et al: Modification of γ-secretase by
nitrosative stress links neuronal ageing to sporadic Alzheimer's
disease. EMBO Mol Med. 4:660–673. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Murphy K, Park AJ, Hao Y, Brewer D, Lam JS
and Khursigara CM: Influence of O polysaccharides on biofilm
development and outer membrane vesicle biogenesis in Pseudomonas
aeruginosa PAO1. J Bacteriol. 196:1306–1317. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mazzio EA, Bauer D, Mendonca P, Taka E and
Soliman KFA: Natural product HTP screening for attenuation of
cytokine-induced neutrophil chemo attractants (CINCs) and
NO2- in LPS/IFNγ activated glioma cells. J
Neuroimmunol. 302:10–19. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Mateo J, García-Lecea M, Cadenas S,
Hernández C and Moncada S: Regulation of hypoxia-inducible
factor-1alpha by nitric oxide through mitochondria-dependent and
-independent pathways. Biochem J. 376:537–544. 2003.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Koivunen P, Hirsilä M, Remes AM, Hassinen
IE, Kivirikko KI and Myllyharju J: Inhibition of hypoxia-inducible
factor (HIF) hydroxylases by citric acid cycle intermediates:
Possible links between cell metabolism and stabilization of HIF. J
Biol Chem. 282:4524–4532. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Goetzl EJ, Huang MC, Kon J, Patel K,
Schwartz JB, Fast K, Ferrucci L, Madara K, Taub DD and Longo DL:
Gender specificity of altered human immune cytokine profiles in
aging. FASEB J. 24:3580–3589. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wiesner D, Merdian I, Lewerenz J, Ludolph
AC, Dupuis L and Anke Witting A: Fumaric acid esters stimulate
astrocytic VEGF expression through HIF-1α and Nrf2. PLoS One.
8(e76670)2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fox HC, D'Sa C, Kimmerling A, Siedlarz KM,
Tuit KL, Stowe R and Sinha R: Immune system inflammation in cocaine
dependent individuals: Implications for medications development.
Hum Psychopharmacol. 27:156–166. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Moreira FP, Medeiros JR, Lhullier AC,
Souza LD, Jansen K, Portela LV, Lara DR, da Silva RA, Wiener CD and
Oses JP: Cocaine abuse and effects in the serum levels of cytokines
IL-6 and IL-10. Drug Alcohol Depend. 158:181–185. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lacagnina MJ, Rivera PD and Bilbo SD:
Glial and neuroimmune mechanisms as critical modulators of drug use
and abuse. Neuropsychopharmacology. 42:156–177. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kohno M, Link J, Dennis LE, McCready H,
Huckans M, Hoffman WF and Loftis JM: Neuroinflammation in
addiction: A review of neuroimaging studies and potential
immunotherapies. Pharmacol Biochem Behav. 179:34–42.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
He J, Yang S and Zhang L: Effects of
cocaine on nitric oxide production in bovine coronary artery
endothelial cells. J Pharmacol Exp Ther. 314:980–986.
2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Winick-Ng W, Leri F and Kalisch BE: Nitric
oxide and histone deacetylases modulate cocaine-induced mu-opioid
receptor levels in PC12 cells. BMC Pharmacol Toxicol.
13(11)2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Dhir A and Kulkarni SK: Nitric oxide and
major depression. Nitric Oxide. 24:125–131. 2011.PubMed/NCBI View Article : Google Scholar
|