Introduction
Members of the IL-1 family of cytokines are key
regulators of inflammatory and innate immunity. IL-36 cytokines
[including IL-36α, IL-36β, IL-36γ and IL-36 receptor antagonist
(IL36Ra)] are members of the IL-1 family (1). IL-36α is particularly expressed in
epithelium, keratinocytes, monocytes/macrophages and αβ and γδ T
lymphocytes (1). Psoriasis and
primary Sjogren's syndrome (pSS) are autoimmune-mediated
inflammatory diseases. A previous study showed that IL-36α was
involved in the development of psoriasis, it had a pro-inflammatory
effect (2). The serum levels of
IL-36α were significantly higher in patients with pSS compared with
those in subjects complaining of dry mouth or eyes, but who did not
meet the American-European Consensus Group criteria for pSS. In
addition, the serum levels of IL-36α were correlated with pSS
disease activity (3).
Systemic lupus erythematosus (SLE) is also a common
autoimmune and inflammatory disease, which is characterized by
complement activation, production of numerous auto-antibodies and
damage to multiple organs and tissues. A recent study reported the
involvement of T helper (Th)17 cells and cytokines in the
pathogenesis of SLE (4). Another
study demonstrated that plasma IL-17 levels were elevated in
patients with new-onset SLE, and a positive correlation was
observed between plasma IL-17 and SLE disease activity index
(SLEDAI) (5). Yang et al
(6) found that IL-17 derived from
patients with active SLE could induce the mRNA expression of
adhesion molecules, and promoted the recruitment of neutrophils. In
addition, IL-36 receptor (IL-36R) signaling appears to be crucial
for the control of the IL-23/IL-17/IL-22 axis (7,8). Carrier
et al (9) demonstrated that
IL-36 cytokines are upregulated by Th17 cytokines, whereas elevated
IL-17 expression was observed in psoriasis. IL-36 signaling
facilitates the pathogenesis of renal tubulointerstitial lesions
through the activation of the NOD-, LRR- and pyrin
domain-containing 3 and the IL-23/IL-17 axis (10). Previous studies found that serum
IL-36 levels were elevated in patients with SLE (11,12).
However, the role of IL-36α in patients with SLE remains to be
fully determined. Therefore, the present study investigated the
contribution of IL-36α in the development of SLE, and explored the
regulatory effect of IL-36α on IL-17 in patients with SLE.
Materials and methods
Study samples
Serum was obtained from patients with SLE (n=60; 55
women and 5 men; median age, 37.5 years, age range, 19-73 years)
and healthy controls (n=29; 25 women and 4 men; median age, 32.0
years, age range, 24-45 years) across two centers: The Department
of Nephrology of The First and Second Affiliated Hospitals of Anhui
Medical University, and the Department of Rheumatology of The
Second Hospital of Anhui Medical University). Patients who had
suffered various types of malignancy or severe inflammation were
excluded from the present study. Healthy controls were selected
from the Health Examination Center of The Second Hospital of Anhui
Medical University. The diagnosis of SLE cases was based on the
American College of Rheumatology 1997 revised criteria for SLE
(13). The SLEDAI was used to assess
disease activity. A SLEDAI score ≥5 was defined as active SLE,
whereas a SLEDAI score <5 was defined as inactive SLE (14). Patients with lupus nephritis were
defined by persistent proteinuria (>0.5 g/24 h) or persistent
hematuria, presence of cellular casts or renal biopsy supporting
(14). All procedures performed in
the present study involving human participants were performed in
accordance with the Ethical Standards of the Ethics Committee for
Human Research at the Second Hospital of Anhui Medical University
as well as in accordance with the 1964 Helsinki Declaration and its
later amendments or comparable ethical standards (15). The present study was approved by the
Ethics Committee for Human Research at The Second Hospital of Anhui
Medical University (approval no.PJ-YX2017-013), and all the
participants provided written informed consent prior to being
enrolled in the study.
Biochemical measurements
The patients were made to fast for 8 h after dinner
in the night. The fasting blood samples were drawn in the morning,
as the blood was also used to measured alanine aminotransferase
levels for patients. Blood samples were also collected for
determination of albumin, creatinine, complement, immunoglobulin
(IgG), erythrocyte sedimentation rate (ESR) and extractable nuclear
antigen polypeptide antibodies [antinuclear antibodies,
double-stranded (ds)DNA antibodies] all of which were routine
examinations. Serum was isolated and stored at -80˚C for measuring
IL-36α, IL-36Ra and IL-17 levels. The IL-36α levels (Cusabio
Biotech; cat. no. CSB-El011617HU), serum IL-36Ra (Cusabio Biotech;
cat. no. CSB-EL011616HU) and IL-17 (R&D Systems, Inc.; cat. no.
D1700) levels were measured using human ELISA kits.
Statistical analysis
Data were analyzed using SPSS version 16 (IBM
Corp.). The data are presented as the mean ± standard deviation for
normally distributed variables, or as the median and interquartile
ranges otherwise. Differences between two groups were assessed
either by an independent samples Student's t-test or by a
Wilcoxon's rank sum test for continuous variables. Correlations
between two variables were assessed by Spearman's rank correlation.
Two-sided P<0.05 was considered to indicate a statistically
significant difference.
Results
In total, 60 patients with SLE, including 47 active
patients and 13 inactive patients, and 29 healthy controls were
enrolled in the present study. All patients and healthy controls
were Chinese. There were no significant differences between
patients with SLE and normal controls in terms of age, sex or
creatinine levels (Table I).
Baseline disease activity (n=60) was highly variable, with SLEDAI
scores ranging from 0-36, and a median of 10.58 patients had
positive ANA, whereas 30 patients with SLE had positive
double-stranded (ds)DNA (Table I).
The majority of patients (75%) received prednisone or an equivalent
glucocorticoid. In total, 40 patients (66.67%) received
glucocorticoids and immunosuppressant (Table I).
 | Table IComparison of clinical and laboratory
parameters between the SLE and control group. |
Table I
Comparison of clinical and laboratory
parameters between the SLE and control group.
| Variable | SLE group, n=60 | Control group,
n=29 | P-value |
|---|
| Anti-double stranded
DNA, -/+ | 30/30 | NA | NA |
| Antinuclear
antibodies, -/+ | 2/58 | NA | NA |
| Albumin, g/l | 31.40±8.99 | 47.49±2.52 | 0.01a |
| Creatinine,
µmol/le | 66.00
(51.25,117.75) | 56.00
(50.00,70.50) | 0.087 |
| Proteinuria, mg/24
he | 660
(237.5-3062.5) | NA | NA |
| C3, g/le | 0.68 (0.44-0.93) | NA | NA |
| C4, g/le | 0.09 (0.05-0.22) | NA | NA |
| IgG, g/le | 14.75
(11.54-19.45) | NA | NA |
| IgA, g/le | 2.48 (1.51-3.28) | NA | NA |
| IgM, g/le | 0.95 (0.75-1.35) | NA | NA |
| Erythrocyte
sedimentation rate, mm/he | 40.00
(22.25-69.25) | NA | NA |
| SLEDAIe | 10.00
(6.00-17.50) | NA | NA |
| Received
glucocorticoidsf, n
(%) | 45(75) | NA | NA |
| Received
glucocorticoids or immunosuppressantsg, n (%) | 40 (66.67) | NA | NA |
| IL-36α,
pg/mle | 50.08
(39.01-108.02) | 25.98
(17.19-32.1) | 0.000c |
| IL-36Ra,
pg/mle | 42.6
(29.73-107.08) | 110.45
(45.72-160.95) | 0.007b |
| IL-17,
pg/mle | 39.68
(33.03-82.44) | 35.13
(32.14-41.25) | 0.036a |
The association between serum IL-36α levels with the
clinical and laboratory parameters of patients with SLE were
analyzed, and the results showed that rash, leukocytes, ESR,
anti-dsDNA, proteinuria and hematuresis were positively associated
with the serum IL-36α levels in patients with SLE (Table II). A negative correlation was
observed between complement 3 and serum IL-36α levels. No
significant associations between IL-36α with any other clinical or
laboratory parameters were observed (P>0.05; Table II).
| Table IIAssociation between plasma IL-36α
levels and clinical and laboratory parameters in the patients with
systemic lupus erythematosus. |
Table II
Association between plasma IL-36α
levels and clinical and laboratory parameters in the patients with
systemic lupus erythematosus.
| Parameters | n | Serum IL-36α level,
pg/ml, median (interquartile range) | P-value |
|---|
| Rash | | | 0.003b |
|
Yes | 36 | 69.35
(42.74-141.75) | |
|
No | 24 | 41.23
(30.58-59.62) | |
| Arthritis | | | 0.382 |
|
Yes | 22 | 53.59
(40.14-140.74) | |
|
No | 38 | 49.83
(32.66-95.08) | |
| Oral ulcer | | | 0.647 |
|
Yes | 13 | 58.68
(32.70-145.68) | |
|
No | 47 | 49.14
(39.11-106.98) | |
| Nervous system
disorder | | | 0.597 |
|
Yes | 4 | 55.96
(21.48-114.11) | |
|
No | 56 | 50.08
(39.44-108.02) | |
| Pleuritis | | | 0.405 |
|
Yes | 10 | 71.10
(40.10-127.37) | |
|
No | 50 | 49.39
(37.95-105.4) | |
| Pericarditis | | | 0.555 |
|
Yes | 6 | 51.34
(39.60-107.32) | |
|
No | 54 | 49.39
(37.95-107.32) | |
| Lymphopenia | | | 0.007b |
|
Yes | 17 | 79.70
(48.73-158.64) | |
|
No | 43 | 44.68
(32.72-77) | |
|
Thrombocytopenia | | | 0.085 |
|
Yes | 24 | 119.61
(58.31-165.61) | |
|
No | 36 | 45.12
(32.35-76.09) | |
| ESR | | | 0.001b |
|
Yes | 33 | 77.0
(43.16-150.60) | |
|
No | 27 | 42.37
(31.24-49.64) | |
|
Hypocomplementemia | | | 0.025a |
|
Yes | 40 | 62.43
(39.59-123.46) | |
|
No | 20 | 43.03
(28.48-56.52) | |
| Anti-double
stranded-DNA positive | | | 0.019a |
|
Yes | 30 | 58.63
(40.57-150.16) | |
|
No | 30 | 43.68
(35.05-75.79) | |
| Proteinuria | | | 0.037a |
|
Yes | 45 | 63.19
(39.66-123.44) | |
|
No | 15 | 42.63
(32.72-53.46) | |
| Hematuresis | | | 0.027a |
|
Yes | 24 | 55.40
(39.01-132.48) | |
|
No | 36 | 44.34
(36.25-87.91) | |
| Antinuclear
antibodies positive | | | 0.538 |
|
Yes | 56 | 51.34
(39.59-108.02) | |
|
No | 4 | 36.28
(31.61-182.15) | |
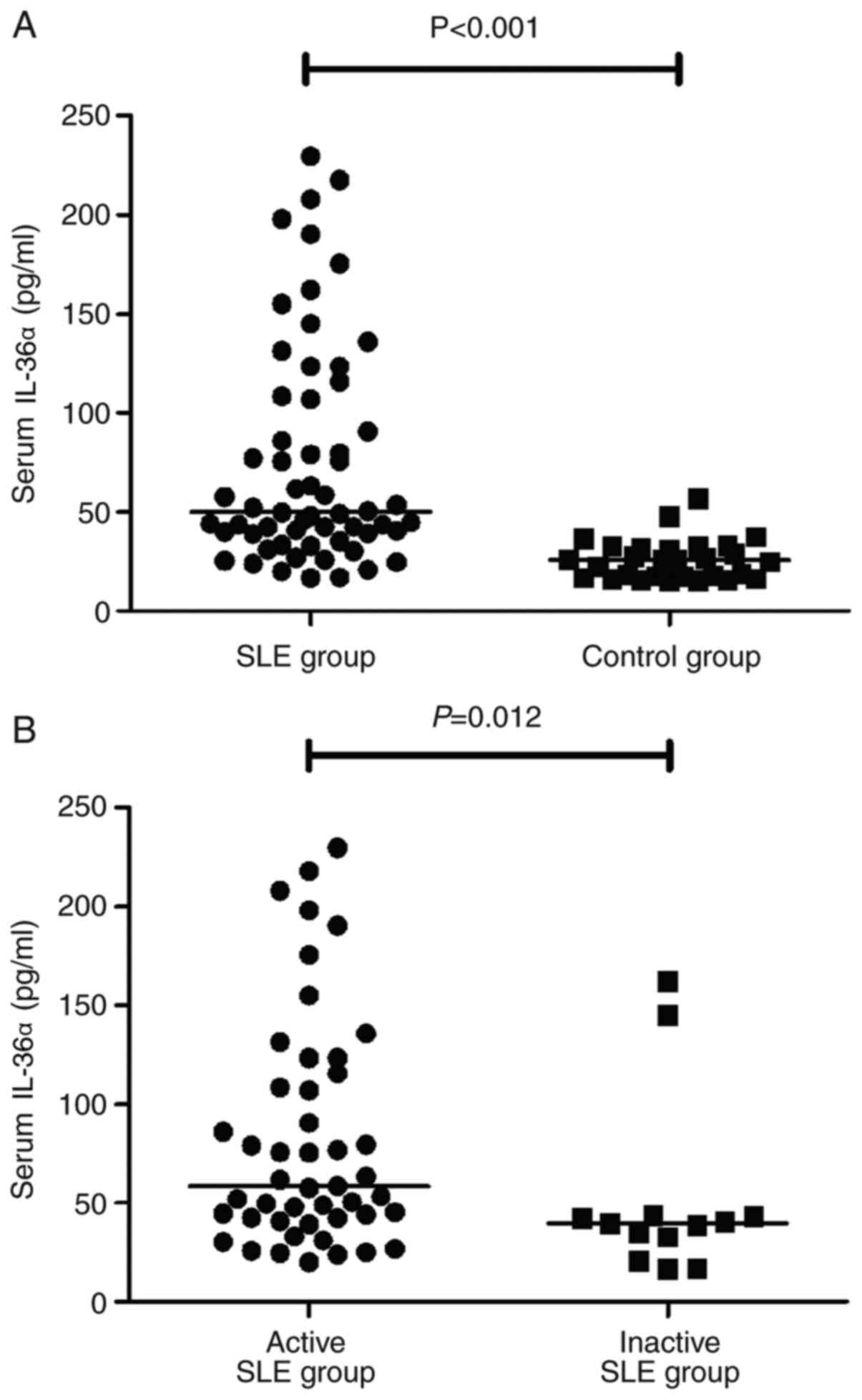
Serum IL-36α levels were significantly higher in
patients with SLE than in healthy controls [median (interquartile
range); 50.08 (39.01-108.02) pg/ml vs. 25.98 (17.19-32.1) pg/ml;
P<0.001)], as shown in Fig. 1A.
The serum IL-36α levels were significantly higher in the active
group compared with those in the inactive group (P=0.012), as shown
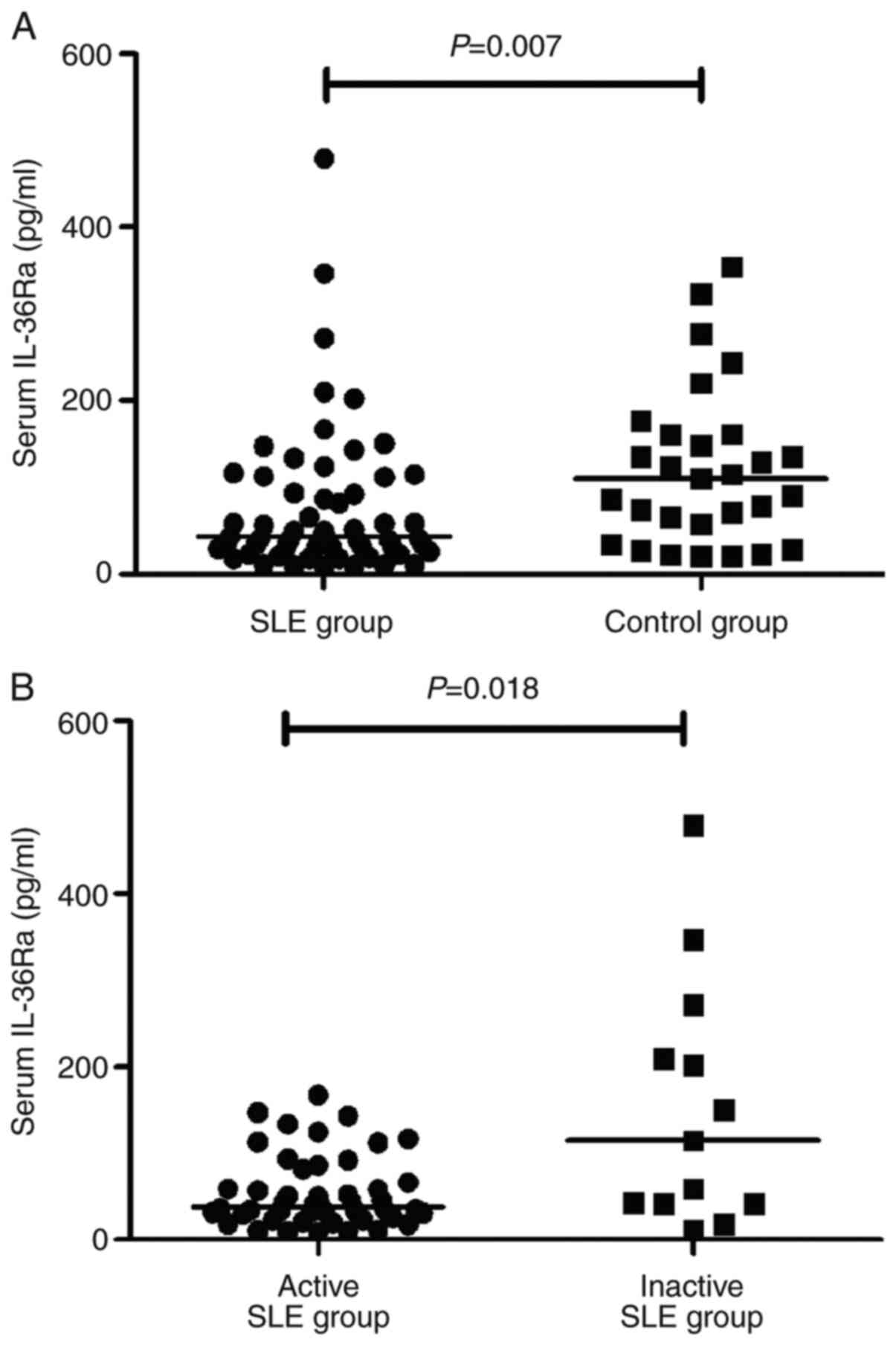
in Fig. 1B. Serum IL-36Ra levels
were significantly lower in patients with SLE than in controls
[42.6 (29.73-107.08) pg/ml vs. 110.45 (45.72-160.95) pg/ml,
P=0.007)], as shown in Fig. 2A, and
it was lower in patients with active SLE compared with the levels
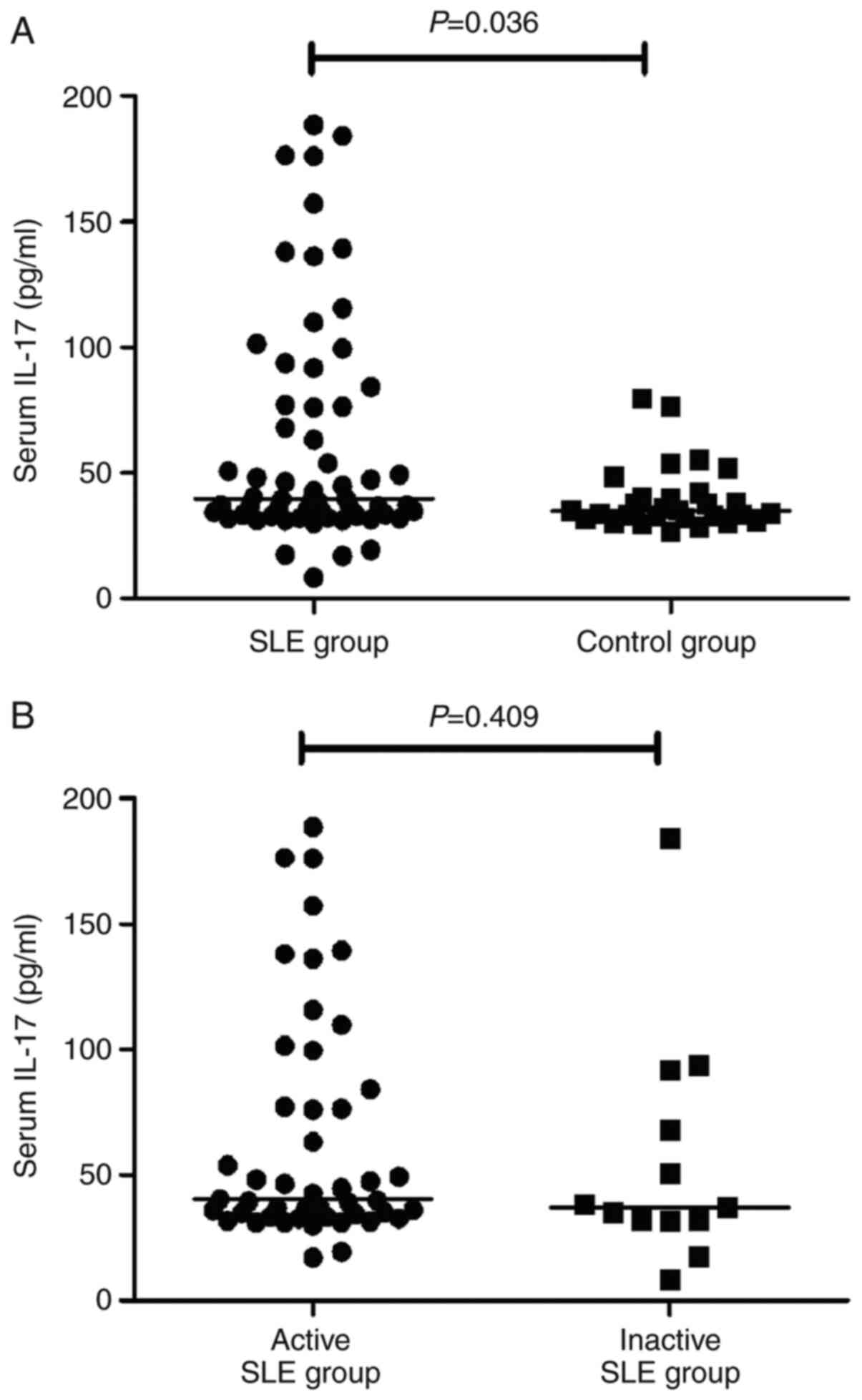
found in patients with inactive SLE (P=0.018, Fig. 2B). Serum IL-17 levels were increased
in patients with SLE (Fig. 3), but
there was no significant difference between patients with active
and inactive SLE.
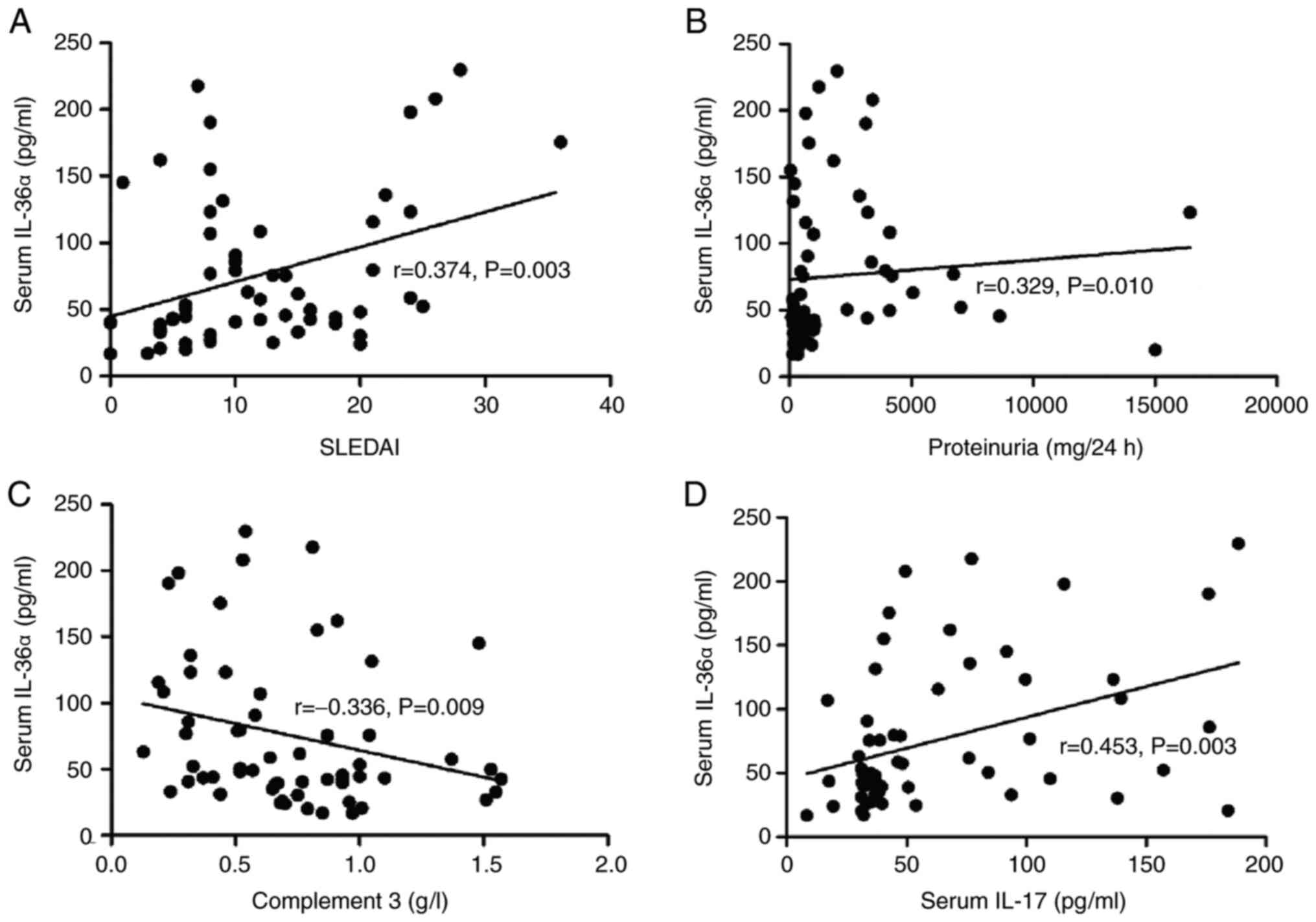
To determine the association between serum IL-36α
levels and disease activity, the association between these two
parameters was analyzed. A positive correlation between serum
IL-36α and the SLEDAI score was observed (r=0.374, P=0.003). In the
correlation analysis shown in Fig.
4, the concentration of serum IL-36α was positively correlated
with proteinuria (r=0.329, P=0.010).
The present study investigated the association
between IL-36α levels with serum IL-17 levels. Serum IL-36α was
positively correlated with IL-17 (r=0.453, P=0.003; Fig. 4). Serum IL-36α levels showed an
inverse correlation with complement 3 (r=-0.336, P=0.009; Fig. 4). There was no significant
correlation between serum IL-36α and IgG, IgA, IgE, IgM, complement
4 or ESR (data not shown).
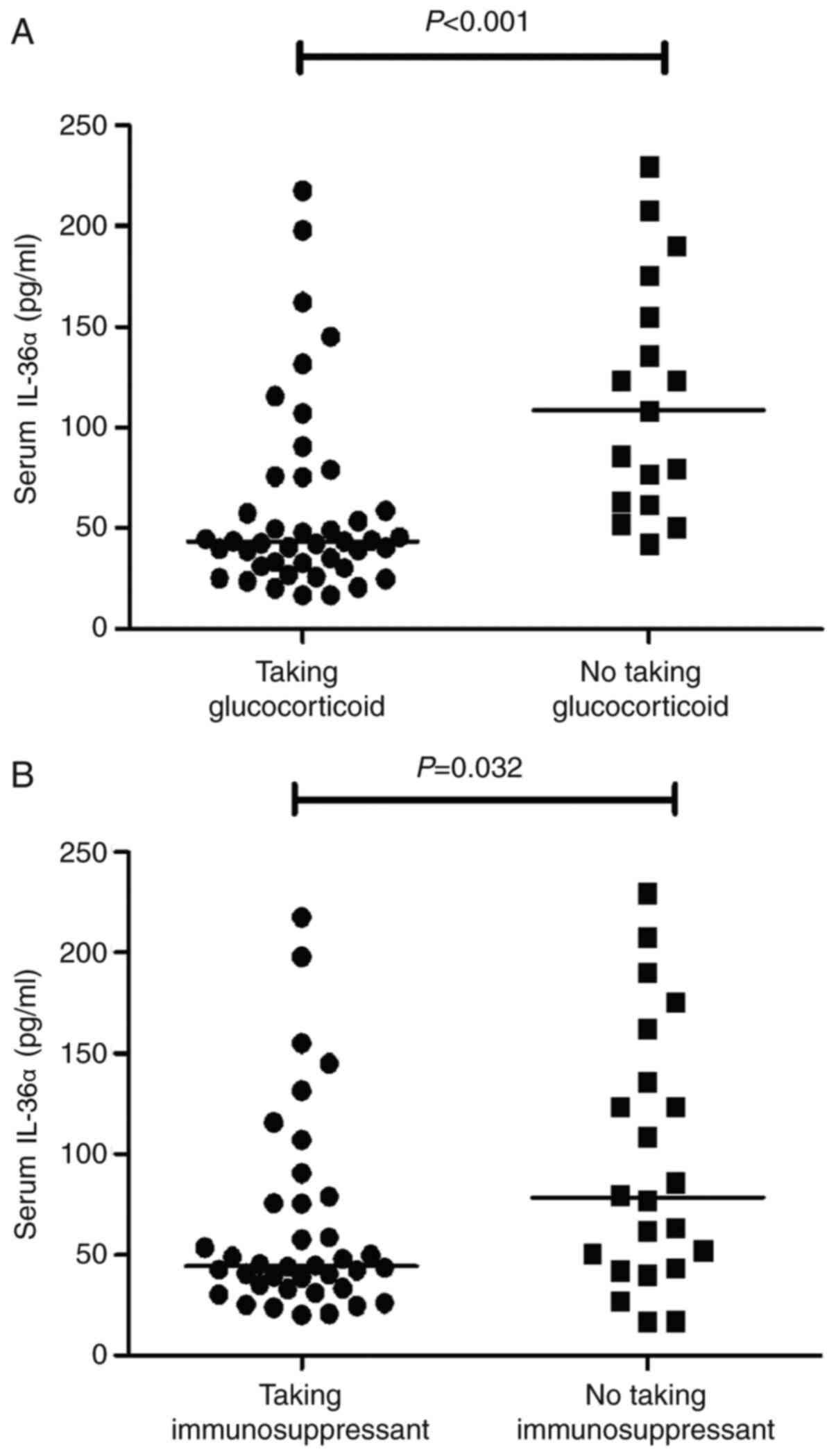
In total, 45 patients received glucocorticoid
treatment, whereas 40 patients with SLE received immunosuppressant
and glucocorticoid therapies. Patients (n=45) who had received
glucocorticoid treatment had lower IL-36α levels compared with
those of patients who had not received glucocorticoid treatment
(P=0.003; Fig. 5). These results
suggested that immunosuppressant and glucocorticoid therapies may
have an inhibitory effect on IL-36α. A total of 47 patients
(78.33%) had renal involvement. Patients with nephritis had higher
serum IL-36α levels than those without nephritis (P=0.037; Table II). Serum IL-36Ra levels were
significantly lower in patients with SLE than in controls. IL-36Ra
levels were negatively correlated with SLEDAI (r=-0.360, P=0.001).
There was no significant association between IL-36Ra levels and
proteinuria, ESR, C3 or C4.
Discussion
A previous study demonstrated that IL-36α binds to a
functional heterodimeric receptor complex, which consists of
IL-36R, IL-1 receptor (IL-1R)-related protein 2 and IL-1R accessory
protein, and activates NF-κB and MAPK to initiate pro-inflammatory
pathways (16).
IL-36α is expressed in epithelium and keratinocytes.
A previous study indicated that IL-36α may be involved in the
pathogenesis of autoimmune disease (2). Previous studies reported that IL-36α
exerts pro-inflammatory effects in the skin of patients with
psoriasis, who exhibit autoimmune-mediated inflammatory skin
lesions (2,17). IL-36 is upregulated in skin lesions
caused by psoriasis (18,19). Moreover, transgenic mice
overexpressing IL-36α in keratinocytes showed certain similarities
to human psoriasis (20).
Although the pathogenesis of SLE is not fully
understood, cytokine-mediated immunity plays an important role in
SLE (4,5). Currently, little is known regarding the
association between IL-36α and SLE. The results of thew present
study showed that the serum IL-36α levels were significantly higher
in patients with SLE compared with those of the controls, and the
serum IL-36α levels were increased in the active SLE group compared
with those of the inactive group. Circulating IL-36α levels were
correlated with SLEDAI, complement 3 and ESR. These results
suggested that IL-36 may be involved in the pathogenesis and
progression of SLE.
Studies have shown that glucocorticoids downregulate
the serum levels of IL-12 family cytokines and IL-37 (21,22). The
most important mechanism by which glucocorticoids exert their
anti-inflammatory effects is considered to be the inhibition of the
NF-κB activity (21,22). In the present study, patients who
were receiving glucocorticoid or immunosuppressant therapy had
lower IL-36α levels, indicating that glucocorticoids or
immunosuppressants may inhibit the production of IL-36α. Zhang
et al (23) found that serum
IL-36 levels in patients with SLE did not differ significantly from
that of the controls. There are several possible reasons for such
inconsistent results. Firstly, the patients in the current study
were primarily in the active state. Furthermore, IL-36α rather than
all IL-36 cytokines serve an important role in autoimmune diseases
(9,24).
A positive correlation between serum IL-36α levels
and proteinuria was observed in the present study. Patients with
kidney involvement had higher serum IL-36 levels. In agreement with
the present study, overexpression of IL-36α (IL-1F6) in the kidney
led to increased expression of IL-6, TGF-β receptor-1 and
mesenchymal markers, and aggravated tumor-infiltrating lymphocytes
in a B6.MRLc1 model (25). In
addition, overexpression of IL-36α upregulated α-smooth muscle
actin expression. Further studies are required to investigate the
expression of IL-36α in lupus nephritis.
IL-17 secretion is primarily derived from Th17
cells, which are CD4+ Th cells that produce IL-17, but
not Th1 or Th2 cytokines. Several studies have reported elevated
serum IL-17a levels in patients with SLE compared with the levels
found in healthy controls (26,27).
Although it was not previously reported, the strong correlation
between serum IL-36α and IL-17 levels that was found in the present
study was expected, since IL-36α acts as a pro-inflammatory
cytokine, upregulating pro-inflammatory mediators, such as IL-6,
IL-8 and TNF-α, and, in combination with TGF-β, may induce naive T
cells to differentiate into Th17 cells (28-31).
This observation suggests that IL-36α may affect the Th17 cell
response in patients with lupus.
IL-36Ra, which is an endogenous receptor antagonist,
displays 50% amino acid sequence homology with IL-1Ra (32). IL-36Ra binds to IL-36R, but does not
induce any cellular response. It prevents the interaction of
IL-36α, IL-36β and IL-36γ with IL-36R, and thus acts as a
endogenous inhibitor (33). In the
present study, serum IL-36Ra levels were lower in patients with SLE
than in the controls. The results of the present study support the
hypothesis that the underlying mechanism of IL-36Ra deficiency
leading to the progression of SLE results from the reduced
suppressing effect of IL-36Ra on IL-36α responses. This imbalance
of IL-36α and IL-36Ra would result in a pathologically increased
Th17 response, which is associated with SLE.
The present study recruited 60 patients with SLE and
29 healthy subjects as the control group. This was a relatively
small sample size, which may be considered a limitation of the
present study. Larger sample sizes are being recruited/collected
for more accurate and representative comparisons in future
studies.
In conclusion, the present study confirmed that
IL-36α was associated with SLE and may be involved in the
regulation of Th17 cytokines. Antagonism of IL-36α should be
explored as a treatment of SLE. Lower IL-36Ra levels resulted in a
reduced inhibitory effect on IL-36α and contributed to SLE
development.
Acknowledgements
Not applicable.
Funding
This study was supported by funding from the Natural Science
Foundation of Anhui Province (grant no. 1508085MH148), the China
Postdoctoral Science Foundation funded project (grant no.
2012M511399) and the Clinical Research Cultivation Program of The
Second Hospital of Anhui Medical University Foundation (grant no.
2020LCYB06).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
DGW conceived and designed the study. XRW and JPX
performed the experiments and wrote the manuscript. All authors
have read and approved the final manuscript. DGW, XRW and JPX
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in the present study
involving human participants were performed in accordance with the
Ethical Standards of the Ethics Committee for Human Research at the
Second Hospital of Anhui Medical University as well as in
accordance with the 1964 Helsinki Declaration and its later
amendments or comparable ethical standards. The present study was
approved by the Ethics Committee for Human Research at The Second
Hospital of Anhui Medical University (approval no.PJ-YX2017-013),
and all the participants provided written informed consent prior to
being enrolled in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van de Veerdonk FL and Netea MG: New
insights in the immunobiology of IL-1 family members. Front
Immunol. 4(167)2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tortola L, Rosenwald E, Abel B, Blumberg
H, Schäfer M, Coyle AJ, Renauld JC, Werner S, Kisielow J and Kopf
M: Psoriasiform dermatitis is driven by IL-36-mediated
DC-keratinocyte crosstalk. J Clin Invest. 122:3965–3976.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Ciccia F, Accardo-Palumbo A, Alessandro R,
Alessandri C, Priori R, Guggino G, Raimondo S, Carubbi F, Valesini
G, Giacomelli R, et al: Interleukin-36α axis is modulated in
patients with primary Sjögren's syndrome. Clin Exp Immunol.
181:230–238. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Perry D, Peck AB, Carcamo WC, Morel L and
Nguyen CQ: The current concept of T(h)17 cells and their expanding
role in systemic lupus erythematosus. Arthritis (Egypt).
2011(810649)2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chen XQ, Yu YC, Deng HH, Sun JZ, Dai Z, Wu
YW and Yang M: Plasma IL-17A is increased in new-onset SLE patients
and associated with disease activity. J Clin Immunol. 30:221–225.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang
X, Wan L and Li M: Th17 and natural Treg cell population dynamics
in systemic lupus erythematosus. Arthritis Rheum. 60:1472–1483.
2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gresnigt MS and van de Veerdonk FL:
Biology of IL-36 cytokines and their role in disease. Semin
Immunol. 25:458–465. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gresnigt MS, Rösler B, Jacobs CW, Becker
KL, Joosten LA, van der Meer JW, Netea MG, Dinarello CA and van de
Veerdonk FL: The IL-36 receptor pathway regulates Aspergillus
fumigatus-induced Th1 and Th17 responses. Eur J Immunol.
43:416–426. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Carrier Y, Ma HL, Ramon HE, Napierata L,
Small C, O'Toole M, Young DA, Fouser LA, Nickerson-Nutter C,
Collins M, et al: Inter-regulation of Th17 cytokines and the IL-36
cytokines in vitro and in vivo: Implications in psoriasis
pathogenesis. J Invest Dermatol. 131:2428–2437. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY,
Hsu YJ, Ka SM, Tsai YL, Liu FC and Chen A: IL-36 signaling
facilitates activation of the NLRP3 inflammasome and IL-23/IL-17
axis in renal inflammation and fibrosis. J Am Soc Nephrol.
28:2022–2037. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mai SZ, Li CJ, Xie XY, Xiong H, Xu M, Zeng
FQ, Guo Q and Han YF: Increased serum IL-36α and IL-36γ levels in
patients with systemic lupus erythematosus: Association with
disease activity and arthritis. Int Immunopharmacol. 58:103–108.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chu M, Wong CK, Cai Z, Dong J, Jiao D, Kam
NW, Lam CW and Tam LS: Elevated expression and pro-inflammatory
activity of IL-36 in patients with systemic lupus erythematosus.
Molecules. 20:19588–19604. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hochberg MC: Updating the American College
of Rheumatology revised criteria for the classification of systemic
lupus erythematosus. Arthritis Rheum. 40(1725)1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bombardier C, Gladman DD, Urowitz MB,
Caron D and Chang CH: Derivation of the SLEDAI. A disease activity
index for lupus patients. The Committee on Prognosis Studies in
SLE. Arthritis Rheum. 35:630–640. 1992.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gandevia B and Tovell A: Declaration Of
Helsinki. Med J Aust. 2:320–321. 1964.PubMed/NCBI
|
|
16
|
Towne JE, Garka KE, Renshaw BR, Virca GD
and Sims JE: Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal
through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to
NF-kappaB and MAPKs. J Biol Chem. 279:13677–13688. 2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Frey S, Derer A, Messbacher ME, Baeten DL,
Bugatti S, Montecucco C, Schett G and Hueber AJ: The novel cytokine
interleukin-36α is expressed in psoriatic and rheumatoid arthritis
synovium. Ann Rheum Dis. 72:1569–1574. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Debets R, Timans JC, Homey B, Zurawski S,
Sana TR, Lo S, Wagner J, Edwards G, Clifford T, Menon S, et al: Two
novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as
an antagonist and agonist of NF-kappa B activation through the
orphan IL-1 receptor-related protein 2. J Immunol. 167:1440–1446.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhou X, Krueger JG, Kao MC, Lee E, Du F,
Menter A, Wong WH and Bowcock AM: Novel mechanisms of T-cell and
dendritic cell activation revealed by profiling of psoriasis on the
63,100-element oligonucleotide array. Physiol Genomics. 13:69–78.
2003.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Blumberg H, Dinh H, Trueblood ES,
Pretorius J, Kugler D, Weng N, Kanaly ST, Towne JE, Willis CR,
Kuechle MK, et al: Opposing activities of two novel members of the
IL-1 ligand family regulate skin inflammation. J Exp Med.
204:2603–2614. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qiu F, Song L, Yang N and Li X:
Glucocorticoid downregulates expression of IL-12 family cytokines
in systemic lupus erythematosus patients. Lupus. 22:1011–1016.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Song L, Qiu F, Fan Y, Ding F, Liu H, Shu
Q, Liu W and Li X: Glucocorticoid regulates interleukin-37 in
systemic lupus erythematosus. J Clin Immunol. 33:111–117.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang M, Xu WD, Zhu Y, Wen PF, Leng RX,
Pan HF and Ye DQ: Serum levels of cytokines in systemic lupus
erythematosus: Association study in a Chinese population. Z
Rheumatol. 73:277–280. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Maddur MS, Miossec P, Kaveri SV and Bayry
J: Th17 cells: Biology, pathogenesis of autoimmune and inflammatory
diseases, and therapeutic strategies. Am J Pathol. 181:8–18.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ichii O, Otsuka S, Sasaki N, Yabuki A,
Ohta H, Takiguchi M, Hashimoto Y, Endoh D and Kon Y: Local
overexpression of interleukin-1 family, member 6 relates to the
development of tubulointerstitial lesions. Lab Invest. 90:459–475.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wong CK, Lit LC, Tam LS, Li EK, Wong PT
and Lam CW: Hyperproduction of IL-23 and IL-17 in patients with
systemic lupus erythematosus: Implications for Th17-mediated
inflammation in auto-immunity. Clin Immunol. 127:385–393.
2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wong CK, Ho CY, Li EK and Lam CW:
Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2
cytokine (IL-4) concentrations in patients with systemic lupus
erythematosus. Lupus. 9:589–593. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Veldhoen M, Hocking RJ, Atkins CJ,
Locksley RM and Stockinger B: TGFbeta in the context of an
inflammatory cytokine milieu supports de novo differentiation of
IL-17-producing T cells. Immunity. 24:179–189. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Nalbandian A, Crispín JC and Tsokos GC:
Interleukin-17 and systemic lupus erythematosus: Current concepts.
Clin Exp Immunol. 157:209–215. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bettelli E, Carrier Y, Gao W, Korn T,
Strom TB, Oukka M, Weiner HL and Kuchroo VK: Reciprocal
developmental pathways for the generation of pathogenic effector
TH17 and regulatory T cells. Nature. 441:235–238. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mangan PR, Harrington LE, O'Quinn DB,
Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR and
Weaver CT: Transforming growth factor-beta induces development of
the T(H)17 lineage. Nature. 441:231–234. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Nicklin MJ: Finally, function for
‘IL-1Fs’. Blood. 118:5713–5714. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Towne JE, Renshaw BR, Douangpanya J,
Lipsky BP, Shen M, Gabel CA and Sims JE: Interleukin-36 (IL-36)
ligands require processing for full agonist (IL-36α, IL-36β, and
IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem.
286:42594–42602. 2011.PubMed/NCBI View Article : Google Scholar
|