Introduction
Bardet-Biedl syndrome (BBS) [Mendelian Inheritance
in Man (MIM), 209900] is a heterogenous disorder that is caused by
the impairment of primary cilia. It belongs to a broad group of
disorders known as ciliopathies, and represents a hallmark exemplar
with a highly variable clinical presentation, likely due to
second-site modification of primary causal loci (1-3).
The predominant clinical features associated with BBS are retinal
degeneration, obesity, polydactyly, cognitive impairment, renal
disease and hypogonadism or urogenital malformations. Minor
symptoms that may complicate a clinical diagnosis of BBS include
developmental delay, behavioral and psychiatric abnormalities,
metabolic and endocrine impairment, cardiovascular involvement,
liver disease, Hirschsprung disease and olfactory deficits
(4). Given the wide phenotypic
variability that exists within and amongst BBS families, a clinical
diagnosis of BBS may prove to be challenging. However, a diagnostic
algorithm has been proposed by the presence of either four major
features, or three major features and two minor symptoms (5). Moreover, it is difficult to make an
accurate early diagnosis since the majority of the symptoms may
only occur over time. Therefore, the median age of diagnosis is 9
years of age, and typically the diagnosis is associated with the
occurrence of retinal degeneration (5,6).
Although certain symptoms can be detected at an antenatal stage,
such as polydactyly or genitourinary abnormalities, in the absence
of a positive family history and established molecular
underpinnings, such a diagnosis is rarely established in early
childhood (7). Obesity, which is
noted in 72-92% of patients with BBS, becomes evident during the
first 3 years of life. Typically, the birth weight is normal, and
the weight gain commences during the first year (5,7).
Obesity is associated with a higher risk of developing diabetes,
metabolic syndrome or hypertension (8,9).
Cognitive difficulties are common (>60% of individuals with
BBS), although only 25% of those observed fulfill the intellectual
disability consensus criteria (10). Some specific deficits, such as
perceptual reasoning, attention capacity and functional
independence, appear to be the most severely affected (10). Other neuropsychiatric abnormalities
have been observed in BBS, including developmental delay, either
motor or language impairment, and a broad spectrum of behavioral
disturbances, such as emotional instability, disinhibition,
aggressiveness, self-injury or obsessive-compulsive behavior
(10). Kidney disease affects
53-82% of patients with BBS, and this represents the common cause
of morbidity and mortality (11).
The renal phenotype is highly variable, with renal dysfunction
leading to end-stage renal failure in 42% of adult patients, as
revealed by a large BBS cohort study (12). Individuals with BBS also display
structural anomalies ranging from cysts, fetal lobulation, renal
dysplasia, calyceal distortion and hydronephrosis to ectopic,
atrophic, horseshoe kidney or renal agenesis (13,14).
Hypogonadism and genital anomalies are observed in 59-98% of
patients. Small penile length has also been identified in nearly
all males with BBS, whereas hypoplastic labia minora is common in
females. Less frequently, hydrometrocolpos may complicate many of
the malformations, including vaginal atresia and septate or
imperforate vagina, which may be identified antenatally or shortly
after birth (5,11,15).
In a minority of individuals, valvular stenosis, atrial/ventricular
septal defects or cardiomyopathy are observed, which may be
diagnosed at the prenatal or neonatal stage (5,16),
whereas anosmia, hearing loss, liver disease, Hirschsprung disease
and laterality defects have been reported at different ages of
onset (4,17,18).
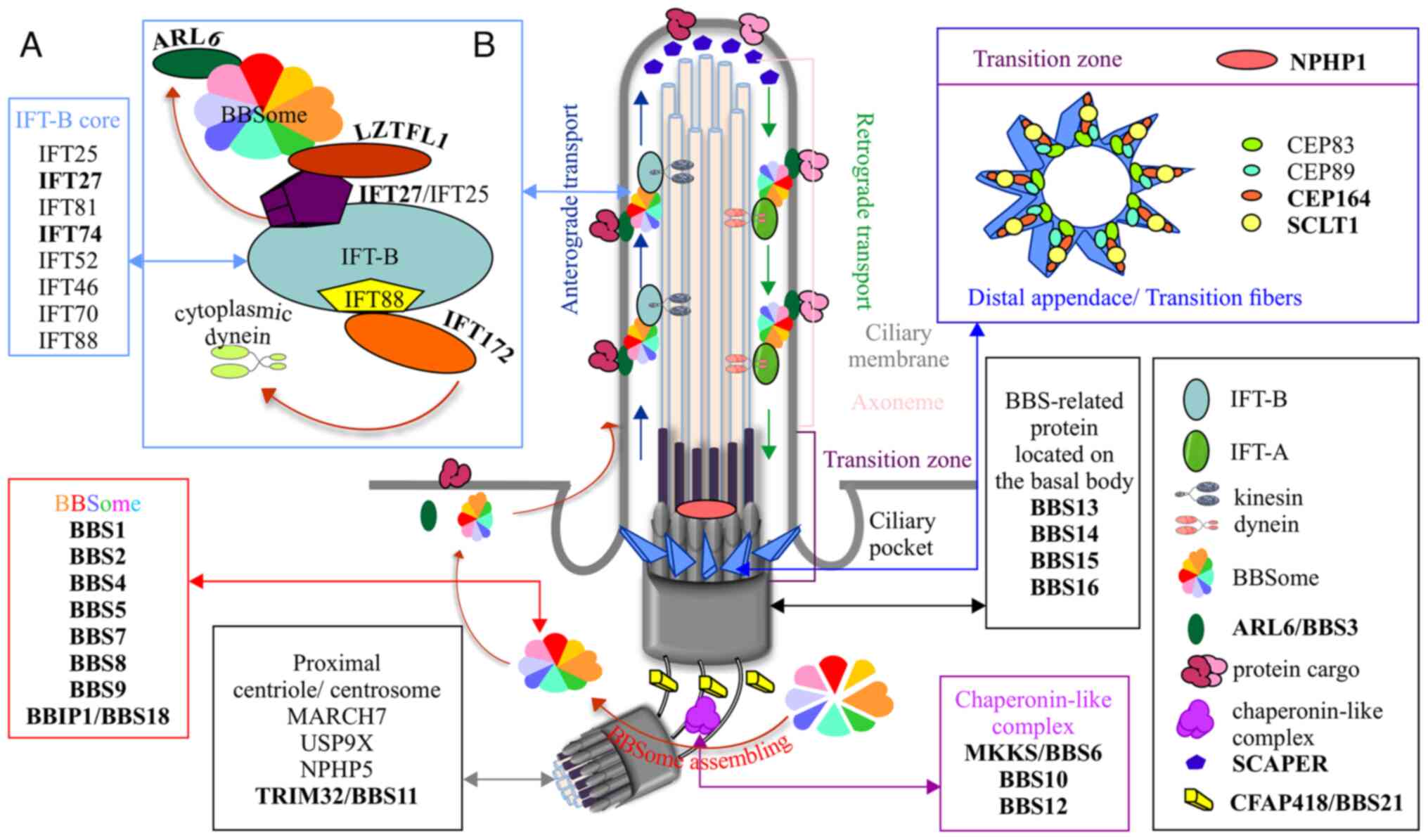
At the time of this report, 26 genes have been
associated with the pathogenesis of BBS (Table I). The majority of the encoded BBS
proteins localize to the base of the cilium, and all have been
shown to be involved in ciliary biogenesis or function (Fig. 1) (11). The BBS1, BBS2, BBS4, BBS5, BBS7,
TTC8/BBS8, BBS9 and BBIP1/BBS18 proteins are components of the
BBSome, a macromolecular complex that functions as an adaptor for
intraflagellar transport (IFT) molecules (19,20).
IFT molecules undergo bidirectional movement along the microtubule
backbone (IFT-A and IFT-B protein complexes), acting as a carrier
for proteins involved either in signaling pathways or in ciliary
homeostasis (21). IFT27/BBS19,
IFT74/BBS20 and IFT172 are components of the IFT-B complex, which
confers anterograde IFT (22).
IFT27/BBS19 has been suggested to interact with ADP ribosylation
factor like GTPase 6 (ARL6)/BBS3, hence modulating the ciliary
export of hedgehog signaling molecules. It has also been proposed
that IFT27/BBS19 may interface with the BBSome complex through an
interaction with leucine zipper transcription factor like 1
(LZTFL1)/BBS17 (23,24). IFT74/BBS20 has been shown to
interact with IFT27/BBS19, whereas the remaining IFT-B molecules,
including IFT172, play an important role in cilium stability
(25,26). The position of the BBSome within the
cilium is stabilized by ARL6/BBS3, a small GTPase that recruits the
BBSome to ciliary membranes (19).
The MKKS centrosomal shuttling protein (MKKS)/BBS6, along with the
BBS10 and BBS12 proteins form part of the chaperonin-like complex
that has an important role in BBSome assembly (27,28).
Several proteins function at the basal body (MKS1/BBS13,
CEP290/BBS14, WDPCP/BBS15 and SDCCAG8/BBS16) and are involved in
ciliogenesis and the modulation of BBSome trafficking within the
ciliary compartment (29-31).
Tripartite motif containing 32/BBS11 is an E3 ubiquitin ligase that
regulates components of the cytoskeleton, whereas LZTFL1/BBS17 is
hypothesized to regulate BBSome activity through transient
interaction with BBS9 (32-34).
Cilia and flagella associated protein (CFAP)418/BBS21 is located at
the base of the cilium, and appears to have a role in facilitating
protein transport, although its complete function and mode of
interaction within the BBS protein network have yet to be fully
elucidated (35). Nephrocystin 1
(NPHP1), localized in the ciliary transition zone, has been shown
to regulate the early stage of cilia formation (31,36)
Recently associated with BBS, sodium channel and clathrin linker 1
(SCLT1) and centrosomal protein (CEP)164 are components of the
distal appendages that are responsible for docking the cilium to
the plasma membrane (37-39).
Both of these are required for ciliary initiation. Another gene
that has recently been shown to be associated with BBS is S-phase
cyclin A associated protein in the ER (SCAPER), which was found to
localize at the ciliary tip, suggesting that it may be involved in
ciliary dynamics during the cell cycle (40). Pathogenic variants in these genes
have been identified in >80% of patients with BBS, and this
percentage has increased rapidly during the past decade due to the
extensive use of next-generation sequencing approaches (7). The most common pathogenic variants
occur in BBS1 and BBS10, accounting for ~45% of
clinically assessed cases. Considered together, the genes that code
for components of the BBSome are most frequently (up to 57%) found
mutated in patients with BBS, and these are followed by the group
of genes that encode chaperonin-like proteins (~30%). Pathogenic
variants identified in ARL6/BBS3 account for ~8% of the clinically
diagnosed individuals. The remainder of the BBS genes are rarely
found to be causal, and these account for ~5% of cases; moreover,
certain variants found in these genes have been reported in only a
few families (41,42). However, the frequency of a specific
pathogenic variant appears to be correlated with the ethnic
background of the affected individuals. Whereas BBS1 and
BBS10 are most frequently impaired amongst individuals of
European descent, pathogenic variants in BBS4, BBS5
and BBS8 appear to be enriched in Middle Eastern and North
African populations (43-45).
Notably, there is a higher prevalence of ARL6/BBS3
pathogenic variants in consanguineous Saudi and Indian families
(46,47).
 | Table ICausal Bardet-Biedl syndrome
genes. |
Table I
Causal Bardet-Biedl syndrome
genes.
| Gene no. | Gene name | Alias | MIM number | Chromosomal
location | Subcellular
location of related proteins |
|---|
| 1 | BBS1 | | 209901 | 11q13.2 | BBSome |
| 2 | BBS2 | RP74 | 606151 | 16q13 | BBSome |
| 3 | ARL6 | BBS3, RP55 | 608845 | 3q11.2 | BBSome
associated |
| 4 | BBS4 | | 600374 | 15q24.1 | BBSome |
| 5 | BBS5 | | 603650 | 2q31.1 | BBSome |
| 6 | MKKS | HMCS, KMS, MKS,
BBS6 | 604896 | 20p12.2 | Chaperonin
complex |
| 7 | BBS7 | | 607590 | 4q27 | BBSome |
| 8 | TTC8 | BBS8, RP51 | 608132 | 14q31.3 | BBSome |
| 9 | PTHB1 | BBS9 | 607968 | 7p14.3 | BBSome |
| 10 | BBS10 | C12orf58,
FLJ23560 | 610148 | 12q21.2 | Chaperonin
complex |
| 11 | TRIM32 | HT2A, LGMDR8,
BBS11 | 602290 | 9q33.1 | Cilium base |
| 12 | BBS12 | FLJ35630,
C4orf24 | 610683 | 4q27 | Chaperonin
complex |
| 13 | MKS1 | MKS, BBS13,
JBTS28 | 609883 | 17q22 | Basal body |
| 14 | CEP290 | KIAA03733H11AG,
JBTS5, SLSN6, LCA10, BBS14 | 610142 | 12q21.32 | Basal body |
| 15 | WDPCP | C2orf86, BBS15,
CHDTHP | 613580 | 2p15 | Basal body |
| 16 | SDCCAG8 | CCCAP, SLSN7,
BBS16 | 613524 | 1q43-q44 | Basal body |
| 17 | LZTFL1 | BBS17 | 606568 | 3p21.31 | BBSome
associated |
| 18 | BBIP1 | NCRNA00081, BBIP10,
BBS18 | 613605 | 10q25.2 | BBSome |
| 19 | IFT27 | RABL4, BBS19 | 615870 | 22q12.3 | IFT |
| 20 | IFT74 | CCDC2, CMG1 | 608040 | 9p21.2 | IFT |
| 21 | CFAP418 | C8orf37, CORD16,
RP64, BBS21 | 614477 | 8q22.1 | Cilium base |
| 22 | NPHP1 | | 607100 | 2q13 | Transition
zone |
| 23 | IFT172 | | 607386 | 2p23.3 | IFT |
| 24 | SCAPER | | 618195 | 15q24.3 | Cilium tip |
| 25 | SCLT1 | | 611399 | 4q28.2 | Distal
appendage |
| 26 | CEP164 | | 614848 | 11q23.3 | Distal
appendage |
BBS has been shown to be predominantly inherited in
an autosomal recessive fashion, although it may also be inherited
as an oligogenic trait (48,49).
The underlying molecular mechanism is often complicated through the
intervention of a third mutant locus, giving rise to ‘triallelic
inheritance’, which may explain the extensive clinical variability
of patients with BBS (50).
Similarly, it has been hypothesized that the presence of
second-site modifier or epistatic interactions are responsible both
for intrafamilial or interfamilial clinical heterogeneity and for
the severity of the phenotype (2,51,52).
Copy number variants and retrotransposon insertions have been
proposed to contribute to the pathogenesis of BBS (53,54).
Furthermore, it has been suggested that even environmental events
may be involved in defining the complexity of the BBS phenotype
(55).
Here, a hitherto unreported case of BBS, clinically
diagnosed in accordance with consensus criteria established by
Beales et al (5), that was
caused by a rare recurrent c.1063C>T; p.Arg355*
variant in BBS12 is described. The molecular finding was
identified by whole exome sequencing (WES) and confirmed by Sanger
sequencing.
Case report
Written informed consent was obtained from the legal
guardian of this patient and her family members, and they were all
enrolled in the research study approved by Institutional Review
Board of University of Medicine and Pharmacy ‘Carol Davila’
Bucharest (approval no. 29700, T.42; Oct 01, 2015). Additionally,
the present study conformed to the guidelines of the Declaration of
Helsinki (56). EDTA-treated
peripheral blood samples from willing family members were collected
(the patient, the patient's sibling and their parents) subsequent
to informed consent. Genomic DNA was extracted from the blood using
the PureLink® Genomic DNA Extraction kit (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. WES was performed by the Advanced Center for
Translational and Genetic Medicine, Ann & Robert H. Lurie
Children's Hospital of Chicago, IL, USA, according to a research
study approved by the Lurie Children's Hospital IRB (approval no.
IRB 2019-3057; Aug 5, 2019). WES was performed on proband genomic
DNA samples according to an established protocol (LC Sciences,
LLC). Fragmented DNA samples generated via sonication were
subjected to library construction. Exome capture was performed
using an Agilent SureSelect Human All Exon V6 kit (Agilent
Technologies, Inc.) according to the manufacturer's instructions,
and next generation sequencing was subsequently performed using an
Illumina Novaseq6000 system at Lianchuan Bio for a 150 bp
paired-end run, to a mean target depth of 147X, generating a total
of 74,060,974 paired-end reads.
For bioinformatics analysis, and prior to alignment,
low-quality reads (first, reads containing sequencing adaptors, and
secondly, nucleotides with a quality score <20) were removed to
yield a total of 73,150,226 cleaned paired-end reads. The
Burrows-Wheeler Aligner (57) was
utilized to perform reference genome alignment (hg19) with reads
contained in paired FASTQ files. As the first post-alignment
processing step, Picard (a collection of command-line tools for
handling high-throughput sequencing data; broadinstitute.github.io/picard/) was utilized to
identify and mark duplicate reads from BAM files. In the second
post-alignment processing step, local read realignment was
performed to correct for potential alignment errors around indels.
Variant calls were generated using GATK HaplotypeCaller (gatk.broadinstitute.org/hc/en-us)
(58) [which calls
single-nucleotide polymorphisms (SNPs) and indels simultaneously
via local de novo assembly of haplotypes in an active
region] or UnifiedGenotyper (59)
(which calls SNPs and indels on a per-locus basis) (60). A Gaussian mixture model was used to
assign accurate confidence scores to each putative variant call,
and SnpEff (pcingola.github.io/SnpEff/) (an open-source tool that
annotates variants and predicts their effects on genes by using an
interval forest approach) was utilized to add biological
information for the variants (61).
Rare variants with gnomAD minor allele frequency <0.01 were
retained, and functional DNA changes impacting amino acid sequence
and intron-exon junctions in the 26 known BBS genes (Table I) were prioritized for further
analysis using the Integrated Genomics Viewer (software.broadinstitute.org/software/igv/home)
(62). BBS12 c.1063C>T;
p.Arg355* was confirmed in the proband and available
family members by PCR amplification with the following
thermocycling conditions: Initial denaturation, 95˚C for 5 min;
followed by 10 cycles of denaturation at 95˚C for 30 sec, annealing
at 66˚C for 30 sec, and extension at 72˚C for 30 sec (-1˚C/cycle);
40 cycles of denaturation at 95˚C for 30 sec, annealing at 56˚C for
30 sec, and extension at 72˚C for 30 sec; and a final extension
step at 72˚C for 10 min. The sequences of the primers used were:
BBS12_PCR1 forward, 5'-TTGTGTGCAACAAGGCAAC-3' and reverse,
5'-TTCACTGAGCCGATTACCAAC-3'. This was followed by capillary
sequencing using BigDye terminator 3.1 chemistry using an ABI
3730xl DNA Analyzer according to the manufacturer's protocols
(Applied Biosystems; Thermo Fisher Scientific, Inc.).
The proband was the first daughter of a young
(mother 17 years-old, father 20 years-old) and apparently healthy
Romani couple. The family self-reported as non-consanguineous. The
second daughter was reported to be healthy. The family history
included Down's syndrome in a paternal cousin, as well as several
(>3 cases) familial cases of intellectual disability on the
father's side of the family. The patient was born at 42 weeks by
vaginal delivery after an uneventful pregnancy. The physical
parameters at birth were within the expected normal range [weight,
3,070 g (70th percentile); length, 50 cm (30th percentile); the
occipitofrontal circumference (OFC) was not provided]. The patient
was admitted to the intensive care unit for 12 h, needing incubator
support due to the poor adaptation. Subsequently, the post-natal
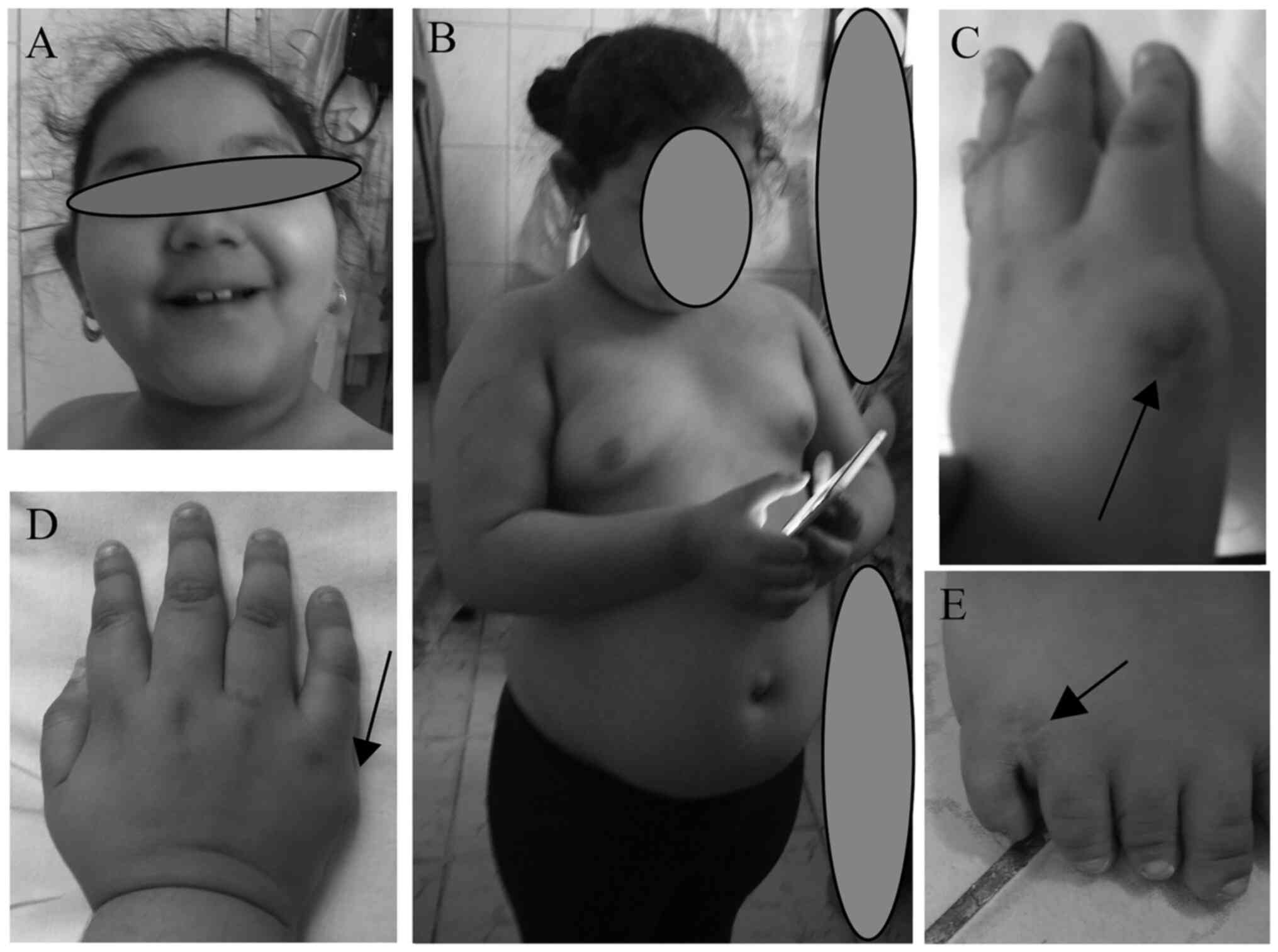
development progressed normally. Postaxial polydactyly was noted in
all four limbs, and supernumerary digits were removed surgically at
8 months (Fig. 2). Psychomotor
development was normal (the patient was able to sit at 6 months;
walk without support at 14 months; said the first syllables at 6
months; and the first words at 12 months). The patient was
evaluated at 6 years of age by a multidisciplinary team, including
a pediatrician, child psychiatrist, child neurologist,
psychologist, clinical geneticist and ophthalmologist. Clinical
workup revealed that she was obese [her weight was 52 kg (>7.5
standard deviations (SD) above the mean)]; she was of tall stature
[her height was 128 cm (>2.5 SD above the mean)]; and relative
macrocephaly was identified [her OFC was 56 cm (>3.6 SD above
the mean)]. Several dysmorphic traits were also observed, namely a
narrow forehead, a decreased bitemporal diameter, sparse eyebrow
hypertelorism, long and smooth philtrum, large ears and full
cheeks. Furthermore, oral/dental abnormalities were identified,
including dysplastic teeth, a high-arched palate and digit
anomalies, such as brachydactyly, conic fingers, partial cutaneous
syndactyly of the second and third toes, and hypoplasia of the
nails were also noted. An ophthalmological examination revealed
retinal dystrophy; the patient's night vision was also very poor,
and her daylight vision was weak (she frequently collides with
objects while walking) as reported by her. However, specific
measurements of visual acuity could not be obtained due to
non-cooperation and severe intellectual disability. The
neurological evaluation revealed language impairment (echolalia,
bradylalia, a limited vocabulary and the use of expressions that
the patient had heard on television) and no sphincter control.
Psychiatric and psychological workup revealed severe intellectual
disability (IQ score 36), behavioral disturbances, including
emotional instability, self-aggressiveness, addictive behavior
towards the phone and television (the patient liked to listen to
music, sing and dance), severe hyperkinesia and abnormal food
behavior (the patient asked repeatedly for food). The patient knew
her name and age, and could count up to 10; however, she could not
recognize colors or play with a puzzle. Abdominal ultrasound
revealed the presence of hypoplastic genitalia, although her liver
and kidneys appeared normal. Likewise, electroencephalography and
brain MRI investigations were unremarkable. Over the course of the
last year (at 7 years of age) slightly elevated levels of
cholesterol [5.4 mmol/l (normal range <5.2 mmol/l)], creatinine
[68 µmol/l (normal range 35-65 µmol/l)] and urea [8.4 mmol/l
(normal range 1.4-8.3 mmol/l)] were recorded for the patient, and
she displayed several episodes of high blood pressure that
responded well to treatment.
Discussion
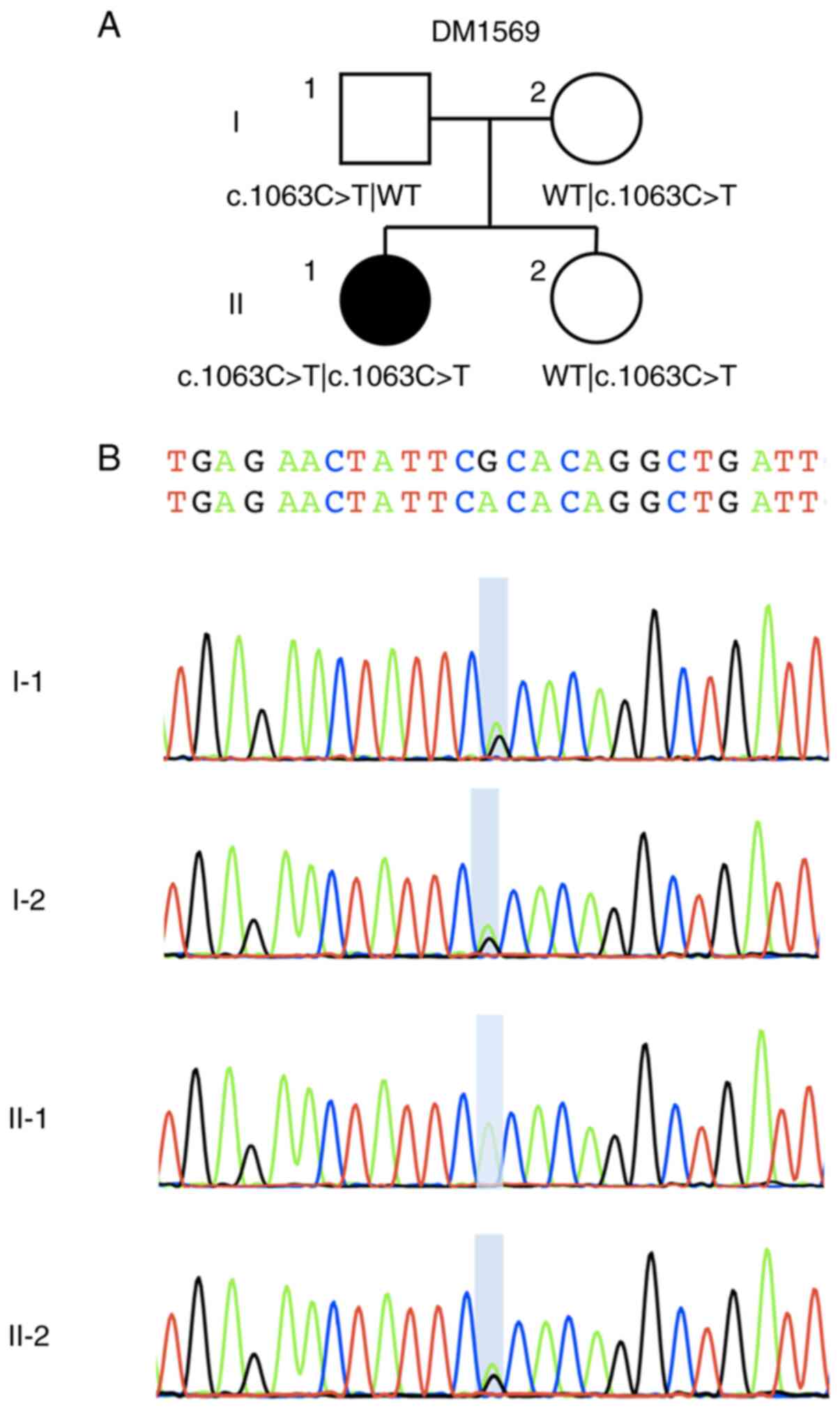
In this case report, a homozygous BBS12
NM_152618.3: c.1063C>T, p.Arg355* variant was
identified using WES. This change was confirmed by Sanger
sequencing and was shown to segregate with disease in the pedigree;
both parents and the unaffected sibling were heterozygous carriers
(Fig. 3). This variant has been
reported previously in dbSNP (rs121918327; ncbi.nlm.nih.gov/snp/), and in ClinVar
(VCV000001147.9; ncbi.nlm.nih.gov/clinvar/) as being pathogenic
according to the American College of Medial Genetics and Genomics
guidelines (63) (PVS1, PP3, PM2).
The variant is a nonsense mutation that is predicted to result in a
premature stop codon within the apical domain of the protein
(64). Experimental validation
remains necessary to determine whether p.Arg355*
produces an unstable protein that is targeted for degradation, or
whether it generates a stable polypeptide with compromised
function.
BBS12 (MIM 610683) is located on chromosome
4q27 and contains two exons, which code for a protein of 710 amino
acids that belongs to a chaperonin-like complex, in addition to
MKKS/BBS6 and BBS10(41). The
chaperonin-like complex, MKKS/BBS6-BBS10-BBS12, was initially
considered to be vertebrate specific, and the proteins have
similarity to the canonical type II chaperonins that are present in
eukaryotic organisms (64-66).
Subsequently, new evidence revealed that the proteins evolved
earlier, due to the presence of several orthologs in ancient
eukaryotes (67). Whereas canonical
eukaryotic chaperonins utilize an ATP-specific hydrolytic site for
protein folding, the rapidly evolved chaperonin-like proteins lost
the ATPase hydrolytic site, but acquired novel functions, including
the transduction of different morphogenetic signals from cilia
(64,67).
The three chaperonin-like proteins have been shown
to be localized at the base of the cilium, in the pericentriolar
region of the basal body and centrosome. They are required for
initial assembly of the BBSome, and operate through stabilizing the
BBS7 protein and subsequently recruiting BBS2 protein as an
intermediary protein for the binding of a six prototypic
chaperonin-containing tailless complex, which is responsible for
completion of the folding process (27). Disruption of one of the
chaperonin-like BBS genes leads to degradation of at least two
subunits of the BBSome. The remaining BBSome proteins either stand
as monomers or form aggregates with unspecified proteins (27).
As a consequence associated with this phenotype, it
has been suggested that the deleterious variants in the
MKKS/BBS6-BBS10-BBS12 complex may lead to a more severe phenotype
and earlier onset of the disease compared with variants in the
BBSome subunits (68,69). This may be accounted for by the
existence of an intermediary complex that manages to retain some
residual function in spite of a BBSome component being impaired,
whereas alteration of the chaperonin-like complex components serve
to restrain the aggregation of any functional complex (28). There is also some evidence to
suggest that visual impairment is most severe in cases associated
with alterations in the chaperonin-like BBS genes, with similar
effects observed for all three genes (68). Furthermore, cognitive impairment is
highly prevalent in individuals with BBS12 variants, whereas
urogenital abnormalities are more common in those carrying
BBS10 pathogenic variants (69).
Four cases with BBS12 c.1063C>T,
p.Arg355*, have been reported previously (Table II) (64,70,71).
In total, 3 of the 5 patients (including the presented case)
reported are Romani; however, the ethnic backgrounds of the other 2
patients have not been provided, so it is not possible to conclude
whether they share the same ethnicity. Interestingly, one of the
patients reported previously is also Romanian, and although he is
not located in the same geographic area as the current patient, the
presence of a putative founder mutation cannot be excluded.
Furthermore, the phenotypes of patients #1 and #2 were not
reported. For the remaining 3 patients, some similarities have been
observed: Genital anomalies were present in all cases. The other
findings are variable, and may be explained either by the young age
of patient #3 at the time of study, given that certain symptoms
occur later in life, or by an additional genomic variant in patient
#4 that may have influenced the phenotype. Even though they
harbored the same variants, polydactyly was noted in all four limbs
in the current patient, whereas in patient #3 polydactyly was
observed only in the feet. The heart anomaly described in patient
#3 was not present in the patient reported here. Therefore, further
studies are required to elucidate the complex pathological
mechanisms underpinning this highly heterogenous ciliopathy.
| Table IIClinical findings of the patients
with BBS harboring the BBS12:c.1063C>T homozygous variant. |
Table II
Clinical findings of the patients
with BBS harboring the BBS12:c.1063C>T homozygous variant.
| Patient
characteristics | Present case | Case #1 and #2 | Case #3 | Case #4 |
|---|
| Reference | - | (57) | (62) | (63) |
| Additional genomic
variants | - | - | - |
BBS1:c.1016A>T |
| Age at time of
report | 7 years | NP | 5 months | NP |
| Sex | Female | NP | Male | Female |
| Ethnic
background | Romani | Romani | NP | NP |
| Phenotype | | | | |
|
Retinitis
pigmentosa | Yes | NP | No | Yes |
|
Obesity | Yes (>6 standard
deviations) | NP | Yes (>97th
percentile) | No |
|
Intellectual
disability | Severe (IQ 36)
(cognitive and language impairment) | NP | No | Yes (cognitive,
language and motor impairment) |
|
Polydactyly | All limbs | NP | Feet | Feet |
|
Genital
anomalies/ hypogonadism | Yes (hypoplastic
genitalia) | NP | Yes (small penis,
small testicles) | Yes (NS) |
|
Kidney
disfunction/ anomaly | Yes (elevated
levels of creatinine and urea) | NP | No | Yes (NS) |
|
Miscellaneous | Severe behavioral
abnormalities, hypercholesterolemia, hypertension, brachydactyly,
syndactyly of 2-3 toes | NP | Heart anomaly,
brachydactyly, syndactyly of 5-6 left toes |
Hypercholesterolemia |
In conclusion, the present case report has provided
novel evidence in terms of defining the phenotype associated with
this rare variant in BBS12.
Acknowledgements
Not applicable.
Funding
This study was funded by the US National Institutes of Health
(grant nos. R01 HD042601, R01 DK072301 and R01 GM121317). EED is
the Ann Marie and Francis Klocke, MD Research Scholar.
Availability of data and materials
Due to constraints of participant consent, whole
exome sequencing data are not posted to public databases, but we
will make portions of the dataset available to researchers upon
reasonable request.
Authors' contributions
IOF collected the data, wrote the manuscript, and
prepared the figures and the tables. MBu and CB provided the
clinical care of the patient. SK performed Sanger confirmation and
segregation analysis. AS assisted with organization of clinical
samples and data. LCB facilitated the initial preparation of
samples. EED conducted the genetic testing and edited the
manuscript. MBa designed the study, and revised the manuscript. All
authors have read and approved the final manuscript. SK and EED
confirm the authenticity of all the raw sequencing data.
Ethics approval and consent to
participate
Willing family members were enrolled in the PhD
research study approved by Institutional Board of University of
Medicine and Pharmacy ‘Carol Davila’ Bucharest, (approval no.
29700, T.42; Oct 01, 2015), and all experiments conformed with the
guidelines of the Declaration of Helsinki. The use of whole exome
sequencing was approved by the Institutional Review Board of the
Ann & Robert H. Lurie Children's Hospital, Chicago (approval
no. IRB 2019-3057, August 5, 2019).
Patient consent for publication
Written informed consent was obtained from legal
guardians of the patient included in the study for genetic testing
and publication of data and images, as well from all the other
participants from whom samples were obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zaghloul NA and Katsanis N: Mechanistic
insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin
Invest. 119:428–437. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Kousi M, Soylemez O, Ozanturk A, Mourtzi
N, Akle S, Jungreis I, Muller J, Cassa CA, Brand H, Mokry JA, et
al: Evidence for secondary-variant genetic burden and non-random
distribution across biological modules in a recessive ciliopathy.
Nat Genet. 52:1145–1150. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Focșa IO, Budișteanu M and Bălgrădean M:
Clinical and genetic heterogeneity of primary ciliopathies
(Review). Int J Mol Med. 48(176)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Forsythe E and Beales PL: Bardet-Biedl
syndrome. Eur J Hum Genet. 21:8–13. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Beales PL, Elcioglu N, Woolf AS, Parker D
and Flinter FA: New criteria for improved diagnosis of Bardet-Biedl
syndrome: Results of a population survey. J Med Genet. 36:437–446.
1999.PubMed/NCBI
|
|
6
|
Weihbrecht K, Goar WA, Pak T, Garrison JE,
DeLuca AP, Stone EM, Scheetz TE and Sheffield VC: Keeping an eye on
bardet-biedl syndrome: A Comprehensive review of the role of
bardet-biedl syndrome genes in the eye. Med Res Arch.
5(10.18103/mra.v5i9.1526)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Forsythe E, Kenny J, Bacchelli C and
Beales PL: Managing bardet-biedl syndrome-now and in the future.
Front Pediatr. 6(23)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Imhoff O, Marion V, Stoetzel C, Durand M,
Holder M, Sigaudy S, Sarda P, Hamel CP, Brandt C, Dollfus H and
Moulin B: Bardet-Biedl syndrome: A study of the renal and
cardiovascular phenotypes in a French cohort. Clin J Am Soc
Nephrol. 6:22–29. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mujahid S, Hunt KF, Cheah YS, Forsythe E,
Hazlehurst JM, Sparks K, Mohammed S, Tomlinson JW, Amiel SA,
Carroll PV, et al: The endocrine and metabolic characteristics of a
large bardet-biedl syndrome clinic population. J Clin Endocrinol
Metab. 103:1834–1841. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kerr EN, Bhan A and Heon E: Exploration of
the cognitive, adaptive and behavioral functioning of patients
affected with Bardet-Biedl syndrome. Clin Genet. 89:426–433.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Forsyth RL and Gunay-Aygun M: Bardet-biedl
syndrome overview. In: GeneReviews® [Internet]. Adam MP,
Ardinger HH, Pagon RA, et al (eds). University of
Washington, Seattle, WA, 1993.
|
|
12
|
Forsythe E, Sparks K, Best S, Borrows S,
Hoskins B, Sabir A, Barrett T, Williams D, Mohammed S, Goldsmith D,
et al: Risk factors for severe renal disease in bardet-biedl
syndrome. J Am Soc Nephrol. 28:963–970. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Putoux A, Attie-Bitach T, Martinovic J and
Gubler MC: Phenotypic variability of Bardet-Biedl syndrome:
Focusing on the kidney. Pediatr Nephrol. 27:7–15. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Marchese E, Ruoppolo M, Perna A, Capasso G
and Zacchia M: Exploring key challenges of understanding the
pathogenesis of kidney disease in bardet-biedl syndrome. Kidney Int
Rep. 5:1403–1415. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Moore SJ, Green JS, Fan Y, Bhogal AK,
Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JC,
et al: Clinical and genetic epidemiology of Bardet-Biedl syndrome
in Newfoundland: A 22-year prospective, population-based, cohort
study. Am J Med Genet A. 132A:352–360. 2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Elbedour K, Zucker N, Zalzstein E, Barki Y
and Carmi R: Cardiac abnormalities in the Bardet-Biedl syndrome:
Echocardiographic studies of 22 patients. Am J Med Genet.
52:164–169. 1994.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Branfield Day L, Quammie C, Héon E, Bhan
A, Batmanabane V, Dai T and Kamath BM: Liver anomalies as a
phenotype parameter of Bardet-Biedl syndrome. Clin Genet.
89:507–509. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Olson AJ, Krentz AD, Finta KM, Okorie UC
and Haws RM: Thoraco-abdominal abnormalities in bardet-biedl
syndrome: Situs inversus and heterotaxy. J Pediatr. 204:31–37.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jin H, White SR, Shida T, Schulz S, Aguiar
M, Gygi SP, Bazan JF and Nachury MV: The conserved Bardet-Biedl
syndrome proteins assemble a coat that traffics membrane proteins
to cilia. Cell. 141:1208–1219. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nachury MV, Loktev AV, Zhang Q, Westlake
CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF,
Sheffield VC and Jackson PK: A core complex of BBS proteins
cooperates with the GTPase Rab8 to promote ciliary membrane
biogenesis. Cell. 129:1201–1213. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lechtreck KF: IFT-cargo interactions and
protein transport in cilia. Trends Biochem Sci. 40:765–778.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bhogaraju S, Engel BD and Lorentzen E:
Intraflagellar transport complex structure and cargo interactions.
Cilia. 2(10)2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liew GM, Ye F, Nager AR, Murphy JP, Lee
JS, Aguiar M, Breslow DK, Gygi SP and Nachury MV: The
intraflagellar transport protein IFT27 promotes BBSome exit from
cilia through the GTPase ARL6/BBS3. Dev Cell. 31:265–278.
2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Eguether T, San Agustin JT, Keady BT,
Jonassen JA, Liang Y, Francis R, Tobita K, Johnson CA, Abdelhamed
ZA, Lo CW and Pazour GJ: IFT27 links the BBSome to IFT for
maintenance of the ciliary signaling compartment. Dev Cell.
31:279–290. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Brown JM, Cochran DA, Craige B, Kubo T and
Witman GB: Assembly of IFT trains at the ciliary base depends on
IFT74. Curr Biol. 25:1583–1593. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bujakowska KM, Zhang Q, Siemiatkowska AM,
Liu Q, Place E, Falk MJ, Consugar M, Lancelot ME, Antonio A, Lonjou
C, et al: Mutations in IFT172 cause isolated retinal degeneration
and Bardet-Biedl syndrome. Hum Mol Genet. 24:230–242.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Seo S, Baye LM, Schulz NP, Beck JS, Zhang
Q, Slusarski DC and Sheffield VC: BBS6, BBS10, and BBS12 form a
complex with CCT/TRiC family chaperonins and mediate BBSome
assembly. Proc Natl Acad Sci USA. 107:1488–1493. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Alvarez-Satta M, Castro-Sanchez S and
Valverde D: Bardet-biedl syndrome as a chaperonopathy: Dissecting
the major role of chaperonin-like BBS proteins (BBS6-BBS10-BBS12).
Front Mol Biosci. 4(55)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dawe HR, Smith UM, Cullinane AR, Gerrelli
D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N,
Attie-Bitach T, et al: The Meckel-gruber syndrome proteins MKS1 and
meckelin interact and are required for primary cilium formation.
Hum Mol Genet. 16:173–186. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Barbelanne M, Hossain D, Chan DP, Peranen
J and Tsang WY: Nephrocystin proteins NPHP5 and Cep290 regulate
BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol
Genet. 24:2185–2200. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Williams CL, Li C, Kida K, Inglis PN,
Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et
al: MKS and NPHP modules cooperate to establish basal
body/transition zone membrane associations and ciliary gate
function during ciliogenesis. J Cell Biol. 192:1023–1041.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Marion V, Stutzmann F, Gerard M, De Melo
C, Schaefer E, Claussmann A, Hellé S, Delague V, Souied E, Barrey
C, et al: Exome sequencing identifies mutations in LZTFL1, a BBSome
and smoothened trafficking regulator, in a family with Bardet-Biedl
syndrome with situs inversus and insertional polydactyly. J Med
Genet. 49:317–321. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Seo S, Zhang Q, Bugge K, Breslow DK,
Searby CC, Nachury MV and Sheffield VC: A novel protein LZTFL1
regulates ciliary trafficking of the BBSome and Smoothened. PLoS
Genet. 7(e1002358)2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Das A, Qian J and Tsang WY: USP9X
counteracts differential ubiquitination of NPHP5 by MARCH7 and
BBS11 to regulate ciliogenesis. PLoS Genet.
13(e1006791)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Heon E, Kim G, Qin S, Garrison JE, Tavares
E, Vincent A, Nuangchamnong N, Scott CA, Slusarski DC and Sheffield
VC: Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum
Mol Genet. 25:2283–2294. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lindstrand A, Davis EE, Carvalho CM,
Pehlivan D, Willer JR, Tsai IC, Ramanathan S, Zuppan C, Sabo A,
Muzny D, et al: Recurrent CNVs and SNVs at the NPHP1 locus
contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum
Genet. 94:745–754. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Morisada N, Hamada R, Miura K, Ye MJ, Nozu
K, Hattori M and Iijima K: Bardet-Biedl syndrome in two unrelated
patients with identical compound heterozygous SCLT1 mutations. CEN
Case Rep. 9:260–265. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shamseldin HE, Shaheen R, Ewida N,
Bubshait DK, Alkuraya H, Almardawi E, Howaidi A, Sabr Y, Abdalla
EM, Alfaifi AY, et al: The morbid genome of ciliopathies: An
update. Genet Med. 22:1051–1060. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang TT, Chong WM, Wang WJ, Mazo G, Tanos
B, Chen Z, Tran TMN, Chen YD, Weng RR, Huang CE, et al:
Super-resolution architecture of mammalian centriole distal
appendages reveals distinct blade and matrix functional components.
Nat Commun. 9(2023)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wormser O, Gradstein L, Yogev Y, Perez Y,
Kadir R, Goliand I, Sadka Y, El Riati S, Flusser H, Nachmias D, et
al: SCAPER localizes to primary cilia and its mutation affects
cilia length, causing Bardet-Biedl syndrome. Eur J Hum Genet.
27:928–940. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Khan SA, Muhammad N, Khan MA, Kamal A,
Rehman ZU and Khan S: Genetics of human Bardet-Biedl syndrome, an
updates. Clin Genet. 90:3–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Niederlova V, Modrak M, Tsyklauri O,
Huranova M and Stepanek O: Meta-analysis of genotype-phenotype
associations in Bardet-Biedl syndrome uncovers differences among
causative genes. Hum mutat. 40:2068–2087. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
M'Hamdi O, Ouertani I, Maazoul F and
Chaabouni-Bouhamed H: Prevalence of bardet-biedl syndrome in
tunisia. J Community Genet. 2:97–99. 2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Farag TI and Teebi AS: High incidence of
Bardet Biedl syndrome among the Bedouin. Clin Genet. 36:463–464.
1989.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Farag TI and Teebi AS: Bardet-Biedl and
laurence-moon syndromes in a mixed arab population. Clin Genet.
33:78–82. 1988.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Abu Safieh L, Aldahmesh MA, Shamseldin H,
Hashem M, Shaheen R, Alkuraya H, Al Hazzaa SA, Al-Rajhi A and
Alkuraya FS: Clinical and molecular characterisation of
Bardet-Biedl syndrome in consanguineous populations: The power of
homozygosity mapping. J Med Genet. 47:236–241. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sathya Priya C, Sen P, Umashankar V, Gupta
N, Kabra M, Kumaramanickavel G, Stoetzel C, Dollfus H and Sripriya
S: Mutation spectrum in BBS genes guided by homozygosity mapping in
an Indian cohort. Clin Genet. 87:161–166. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Katsanis N, Eichers ER, Ansley SJ, Lewis
RA, Kayserili H, Hoskins BE, Scambler PJ, Beales PL and Lupski JR:
BBS4 is a minor contributor to Bardet-Biedl syndrome and may also
participate in triallelic inheritance. Am J Hum Genet. 71:22–29.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
49
|
Katsanis N: The oligogenic properties of
Bardet-Biedl syndrome. Hum Mol Genet. 13:R65–R71. 2004.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Katsanis N, Ansley SJ, Badano JL, Eichers
ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL and
Lupski JR: Triallelic inheritance in Bardet-Biedl syndrome, a
Mendelian recessive disorder. Science. 293:2256–2259.
2001.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Davis EE, Zhang Q, Liu Q, Diplas BH, Davey
LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, et
al: TTC21B contributes both causal and modifying alleles across the
ciliopathy spectrum. Nat Genet. 43:189–196. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Badano JL, Leitch CC, Ansley SJ,
May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S and
Katsanis N: Dissection of epistasis in oligogenic Bardet-Biedl
syndrome. Nature. 439:326–330. 2006.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lindstrand A, Frangakis S, Carvalho CM,
Richardson EB, McFadden KA, Willer JR, Pehlivan D, Liu P,
Pediaditakis IL, Sabo A, et al: Copy-number variation contributes
to the mutational load of bardet-biedl syndrome. Am J Hum Genet.
99:318–336. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Delvallée C, Nicaise S, Antin M, Leuvrey
AS, Nourisson E, Leitch CC, Kellaris G, Stoetzel C, Geoffroy V,
Scheidecker S, et al: A BBS1 SVA F retrotransposon insertion is a
frequent cause of Bardet-Biedl syndrome. Clin Genet. 99:318–324.
2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Shaheen R, Szymanska K, Basu B, Patel N,
Ewida N, Faqeih E, Al Hashem A, Derar N, Alsharif H, Aldahmesh MA,
et al: Characterizing the morbid genome of ciliopathies. Genome
Biol. 17(242)2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
World Medical Association Declaration of
Helsinki. Ethical principles for medical research involving human
subjects. JAMA. 310:2191–2194. 2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Poplin R, Ruano-Rubio V, DePristo M,
Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD,
Levy-Moonshine A, Roazen D, et al: Scaling accurate genetic variant
discovery to tens of thousands of samples. bioRxiv: doi: https://doi.org/10.1101/201178.
|
|
59
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
60
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Stoetzel C, Muller J, Laurier V, Davis EE,
Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda
P, et al: Identification of a novel BBS gene (BBS12) highlights the
major role of a vertebrate-specific branch of chaperonin-related
proteins in Bardet-Biedl syndrome. Am J Hum Genet. 80:1–11.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
65
|
Katsanis N, Beales PL, Woods MO, Lewis RA,
Green JS, Parfrey PS, Ansley SJ, Davidson WS and Lupski JR:
Mutations in MKKS cause obesity, retinal dystrophy and renal
malformations associated with Bardet-Biedl syndrome. Nat Genet.
26:67–70. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
66
|
Stoetzel C, Laurier V, Davis EE, Muller J,
Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, et al:
BBS10 encodes a vertebrate-specific chaperonin-like protein and is
a major BBS locus. Nat Genet. 38:521–524. 2006.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Mukherjee K and Brocchieri L: Ancient
origin of chaperonin gene paralogs involved in ciliopathies. J
Phylogenetics Evol Biol. 1(107)2013.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Billingsley G, Bin J, Fieggen KJ, Duncan
JL, Gerth C, Ogata K, Wodak SS, Traboulsi EI, Fishman GA, Paterson
A, et al: Mutations in chaperonin-like BBS genes are a major
contributor to disease development in a multiethnic Bardet-Biedl
syndrome patient population. J Med Genet. 47:453–463.
2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Castro-Sánchez S, Álvarez-Satta M, Cortón
M, Guillén E, Ayuso C and Valverde D: Exploring genotype-phenotype
relationships in Bardet-Biedl syndrome families. J Med Genet.
52:503–513. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Iurian SI, Arts H, Brunner H and Fintina
D: Bardet-biedl Syndrome-case presentation. Romanian J Pediatrics.
64:289–292. 2015.
|
|
71
|
Manara E, Paolacci S, D'Esposito F, Abeshi
A, Ziccardi L, Falsini B, Colombo L, Iarossi G, Pilotta A, Boccone
L, et al: Mutation profile of BBS genes in patients with
Bardet-Biedl syndrome: An Italian study. Ital J Pediatr.
45(72)2019.PubMed/NCBI View Article : Google Scholar
|