Introduction
Congenital nephrotic syndrome (CNS) is a rare
autosomal recessive disorder. The most common type of CNS is the
Finnish type, a disorder characterized by massive proteinuria
detected at birth, a large placenta, marked edema and radial
dilatation of the proximal tubules (1). The incidence of CNS in Finland is
estimated to be 1 in 8,200 live births, and is considered to be
lower in other countries. A high incidence of CNS has also been
reported in certain regions of the United States, for example,
among the Old Order Mennonites in Lancaster, PA (2). Contributing mutations to CNS mostly
occur in a panel of five genes: nephrin (NPHS1), podocin
(NPHS2), Wilms tumor (WT1), laminin (LAMB2)
and phospholipase C epsilon 1 (PLCE1) (3). Previous research has tended to focus
on mutations in NPHS1, as it is the main gene involved in
CNS of the Finnish type, in an autosomal recessive manner (3,4).
However, other studies showed that whilst NPHS1 gene
mutations can cause CNS in non-Finnish individuals, they are a less
common cause than NPHS2, WT1, LAMB2 and
PLCE1 mutations (3).
Previous studies have reported NPHS1 mutational analysis
suggests that abnormal or inefficient signaling through the
nephrin-podocin complex contributes to podocyte dysfunction and
proteinuria (5). Typically, the
histological lesions of CNS are detected after 3 months of age when
symptoms appear, such as failure to gain weight and edema (1). In this case report, the case of a
newborn with an early diagnosis of Finnish variant CNS, born to a
Guatemalan mother with a NPHS1 variant, is described, and
was determined to be most likely the result of malfunctional DNA
binding sites for transcription factors in the NPHS1
locus.
Case report
A baby boy was delivered vaginally at 35.1 weeks
gestational age, based on a 24.1-week ultrasound, to a 22-year-old
gravida mother from Guatemala. The mother had previously delivered
a child prematurely at 32 weeks via cesarean section who died in a
Guatemalan hospital for unknown reasons within the first month of
life. Her prenatal laboratory tests were unremarkable, and she
denies any illicit drug use or alcohol/tobacco use. The mother had
late, limited prenatal care that began at 24 weeks, with a limited
number of visits. The pregnancy was complicated by membrane rupture
25 h before delivery. Birth weight was 2.030 kg (12th percentile
average size for gestational age), and suture separation was
evident. He was admitted to the Neonatal Intensive Care Unit (NICU)
for prematurity.
On day of life (DOL) 2, the infant's creatinine was
elevated (1.30 mg/dl) and hypoproteinemia (3.1 g/dl) and
hypoalbuminemia (1.2 g/dl) were evident. Findings were attributed
to poor maternal nutrition and expressed breast milk was fortified
with NeoSure® to 27 kcal/ounce. Hypoalbuminemia and
hypoproteinemia persisted. Bicitra (3 mEq/kg/day) administration
was initiated on DOL 9 to correct metabolic acidosis due to a
bicarbonate level of 15 mEq/l. On DOL 16, urinalysis revealed 3+
protein and 3+ glucose. The next day a urine protein to creatinine
ratio of 59.23 was indicative of nephrotic syndrome.
Transthoracic echocardiogram on DOL 9 showed a
patent foramen ovale with a left to right shunt and mild left
ventricular hypertrophy. On further workup for hypertension, renin
and aldosterone levels were found to be elevated at 130 ng/dl and
428 ng/dl, respectively. Due to persistent hypertension (since DOL
1) above the 95th percentile, captopril (0.05 mg/kg/8 h) was
administered beginning on DOL 16. The renal ultrasound was
unremarkable. His urinalysis revealed normal pH, which was
inadequate in the setting of acidosis.
At 3 months of life, thyroid hormone levels and
antithrombin III activity levels started to decrease, which is
typical for this condition (6). The
patient was placed on levothyroxine, and was treated with albumin
infusions, angiotensin-converting enzyme inhibitors (captopril),
and nonsteroidal anti-inflammatory drug (Indomethacin) with
increased dietary protein supplementation starting on DOL 22. His
serum immunoglobulin levels (IgG, IgA, IgM and IgE) were low and
required IVIG treatment through a port-a-cath. He was placed on
oral supplementation for copper and iron. Mild periorbital and
scrotal edema were first observed on DOL 36. With the
identification of the early onset of nephrotic syndrome, genetic
testing was performed to assess possible NPHS1,
NPHS2, WT1, LAMB2 and PLCE1
mutations.
The patient's genomic DNA was analyzed by Athena
Diagnostics via PCR amplification of purified genomic DNA, followed
by Sanger DNA sequencing of the gene's coding region. In addition,
at least 10 bases of intronic DNA on either side of each exon
containing the highly conserved exon-intron splice junctions were
also sequenced. No variants were detected in NPHS2,
WT1, LAMB2 and PLCE1 (Table I). A positive homozygous variant was
detected in NPHS1, mRNA (NCBI Reference Sequence:
NM_004646.3) located at c.3024A>G (Fig. S1). Up-to-date analysis by the
variant scientists of Athena Diagnostics showed NPHS1
c.3024A>G is a synonymous variant of uncertain significance, as
the available data (Score, 4; range 1-7, benign-pathogenic,
respectively) was insufficient to determine the clinical
significance of the variant at this time. ClinVar-NCBI showed two
cases of the same c.3024A>G type with uncertain significance
(Table II); one is an entry of the
reported variant in the present study (variation ID: 429811).
Up-to-date searches for the criteria provided in ClinVar for
variation ID: 429811, NM_004646.3 (NPHS1):c.3024A>G
(p.Arg1008=) showed multiple submissions, the most recent of which
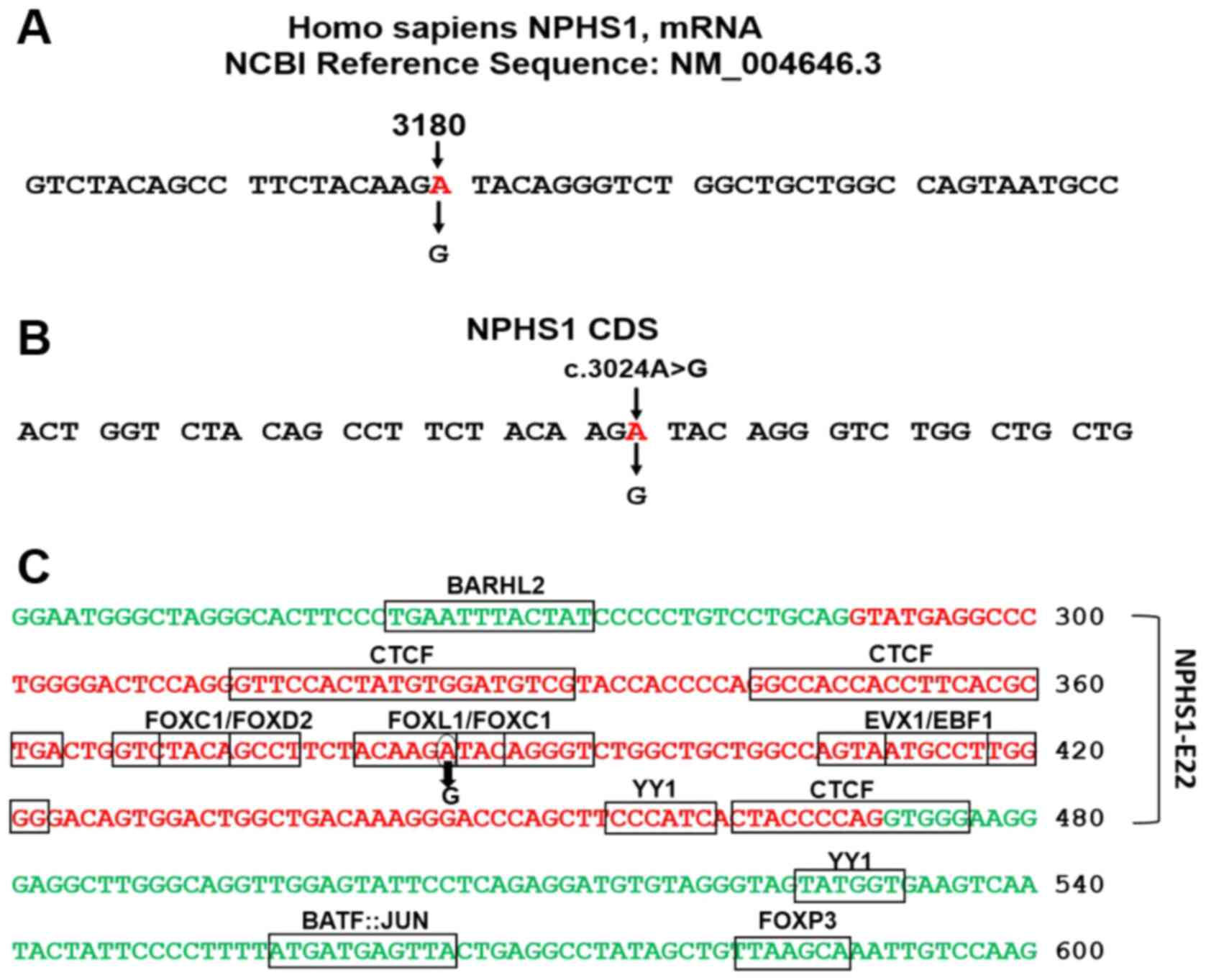
was Sep 26th, 2021. The substitution of A>G occurs in the AGA
codon for arginine; allele A, mapped in the third position of the
triplet at position 3,180 in the NPHS1 mRNA (NM_004646_3)
generating a degenerate redundant AGG codon (Fig. 1A). c.3024 indicates the allele's
position in the coding sequence (Fig.
1B). The variant was identified as a homozygous variant within
NPHS1 exon 22 (Fig. 1C).
There can be little doubt that the CNS case was associated with
this NPHS1 variant. Thus, it was hypothesized that the
generated mutation may cause CNS by disturbing the expression of
NPHS1, via alteration of the regulatory sequences, which is
a likely explanation for the genetic condition. The motifs of the
transcriptional factors binding sites (TFBSs) in NPHS1
exon-22 and adjacent introns' sequences were analyzed using JASPAR
2020 (7,8). The analysis revealed TFBSs presence
for at least 10 types of binding motifs specific to transcription
factors in exon 22 and the neighboring introns' sequences (Fig. 1C). The c.3024A>G variant was
mapped at TFBSs for FOXL1 and FOXC1. A further point is the
c.3024A>G mutation is close to mammalian-wide interspersed
repeats (MIRs), 46 bases away from NPHS1 exon 22 (Fig. S2), the data has been adapted from
Genome Browser UCSC (genome.ucsc.edu/). The main function of MIRs is
exonization, the generation of new exons from intronic DNA
sequences. Additionally, MIRs are considered as major donors of
TFBSs (9,10). Athena Diagnostics computational
tools yielded predictions that this homozygous variant may result
in the gain of a cryptic splice site without affecting the natural
splice sites. Intriguingly, the homozygous variant increased the
double helix stability (Fig. S3)
as measured by oligo calculator in the atdbio website (atdbio.com/tools/oligo-calculator) in
terms of the standard free energy change; such changes are
associated with diseases (11). It
is hypothesized that the child did not have a de novo
mutation, as the family lost their first child, who presented with
a very similar clinical condition. The sequence validation was not
obtainable for the parents, but was likely to be heterozygous for
the NPHS1 variant.
 | Table IPanel of the 5 genes tested for
variants by DNA sequencing in the congenital nephrotic syndrome
case study. Only one variant was identified in the nephrin gene,
NPHS1. |
Table I
Panel of the 5 genes tested for
variants by DNA sequencing in the congenital nephrotic syndrome
case study. Only one variant was identified in the nephrin gene,
NPHS1.
| Gene name:
Description | NCBI gene ID | NCBI nucleotide
ID | OMIM no. | Variant | Clinical
significance |
|---|
| LAMB2: Laminin
subunit β2 | 3913 | NM_002292_3 | OMIM 150325 | No variant
detected | - |
| NPHS1: NPHS1
adhesion molecule, nephrin | 4868 | NM_004646_3 | OMIM 802716 | c.3024A>G | Variant of uncertain
significance |
| NPHS2:
Stomatin family member, podocin | 7827 | NM_014626_3 | OMIM 804768 | No variant
detected | - |
| PLC1:
Phospholipase C γ1 | 5335 | NM_018341_3 | OMIM 808414 | No variant
detected | - |
| WT1:
Transcription factor | 7490 | NM_024428_2 | OMIM 807102 | No variant
detected | - |
| Table IICriteria and submitters of
NPHS1 c.3024 A>G variant in ClinVar. Data adapted from
ClinVar-NCBI. |
Table II
Criteria and submitters of
NPHS1 c.3024 A>G variant in ClinVar. Data adapted from
ClinVar-NCBI.
| Variant location in
coding sequence of NPHS1 mRNA NM_004646.3 | Collection
method | Submitter,
submission date | Clinical
significance | Submission
accession no. |
|---|
| c.3024 A>G | | | | |
|
GRCh37:
Chr19:36330224 | Clinical
testing | GeneDx, May 12,
2017 | Uncertain
significance | SCV000582470.3 |
|
GRCh38:
Chr19:35839322 | | Athena Diagnostics,
Inc., May 8, 2018 | Uncertain
significance | SCV000695728.1 |
Discussion
The present CNS case highlights a rare disease with
an indicator of suspicion regarding the genetic cause of the
disease. Sanger sequencing is an established method and is used for
identification and validation of the presence of the homozygous
mutation; it is unlikely the mutation is de novo, but is
instead from heterozygous parents. Sanger sequencing is used to
identify the pathogenic variation in various diseases (12,13).
Although the patient showed classic symptoms of CNS (1,14), the
detected homozygous c.3024A>G variant produces a degenerate
codon with a genetic condition of uncertain significance. However,
keeping in mind the clinical manifestations associated with CNS and
the fact that no mutations were found in the other four tested
genes associated with CNS (NPHS2, WT1, LAMB2
and PLCE1) (3,14), the potential involvement of
regulatory sequences in NPHS1 exon 22 at the site of
mutation is thus discussed further.
The bioinformatics analysis showed that the A>G
change caused by c.3024A>G mutation could alter transcription
binding sites; specifically, the composition of two motifs specific
to transcription factors FOXL1 and FOXC1. The deregulation of FOX
transcription factors leads to congenital disorders (15,16),
and consequently, these changes may cause potential malfunction of
the transcription process in NPHS1 exon 22.
Intriguingly, previous studies have shown that exons
have regulatory activity in addition to their coding activity. As
early as 1997, the DNase I hypersensitive sites specific DNA
sequences related to the transcriptional activity were identified
in coding exons in mice (17). More
recently, exons have been shown to mediate activation of
transcription starts (18) and have
TFBS sequences (19-21).
Not surprisingly, recent versions of genomics websites show
regulatory sequences spanning coding sequences, for example
NPHS1 ENSG00000161270 and several other genes. Furthermore,
a recent study found that a transcription factor's binding affinity
in exons is weak, but improves in the noncoding sequences of DNA
(22). It is quite possible that
genetic variants may affect exonic splicing regulatory sequences
and consequently disrupt pre-mRNA splicing and initiate genetic
diseases (23,24).
To conclude, a rare NPHS1 gene variant
(25) is described, which likely
caused a disruption in regulatory sequences, TFBSs and cryptic
splice sites (15-17,24,26,27)
associated with CNS. The c.3024A>G variant modified the two
transcription factors', FOXL1 and FOXC1, binding sites in the
NPHS1 exon 22 sequence, and may have influenced MIRs
functions (9,10). Consequently, the mutation is likely
the cause of dysregulated NPHS1 expression. The reported
case should increase awareness of the early diagnosis of CNS in
non-Finish populations. The study may encourage further work to
investigate the clinical significance of the c.3024A>G variant
and potential involvement of exonic splicing regulatory sequences
and other regulatory sequences in genetic conditions of uncertain
significance.
Supplementary Material
Sanger DNA sequencing of the variant
NPHS1 c.3024A>G by Athena Diagnostics. (A) DNA sequence
in a healthy individual, the arrow shows a normal adenine in the
AGA codon for arginine (R). (B) The base pair substitution from A
to G in patient DNA. (C) Chromatogram derived from the analysis of
the NPHS1 c.3024A>G variant. NPHS1, nephrin.
The c.3024A>G mutation is close to
the MIRs, 46 bases away from NPHS1 exon 22. The main
function of MIRs is exonization, the generation of new exons from
intronic DNA sequences. Additionally, MIRs are considered as major
donors of transcription-factor binding sites. The data has been
adapted from Genome Browser UCSC. MIR, mammalian-wide interspersed
repeat; NPHS1, nephrin.
DNA double helix stability of
NPHS1 exon 22 was predicted in normal and mutant sequences
in terms of the standard free energy change: ΔG˚=ΔH˚-TΔS˚. The
double helix stability data was calculated using the oligo
calculator in the atdbio website. ΔG˚, free energy change; ΔH˚,
enthalpy change; TΔS˚, entropy change; NPHS1, nephrin.
Acknowledgements
The authors greatly appreciate the variant's
information provided by Miss Meagan Nashawaty, MS, Certified
Genetic Counselor (CGC), Athena Diagnostics, Marlborough, MA,
USA.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request, and the ClinVar-NCBI repository [ncbi.nlm.nih.gov/clinvar/variation/429811/].
Authors' contributions
TLV analyzed and interpreted the clinical patient
data. MAIAO was responsible for TFBSs, MIRs and homozygous variant
double helix stability analyses. TTT, VGL, DBG, MH and OA cared for
the patient and assisted in writing the case report. TLV and MAIAO
wrote and revised the manuscript. All authors have read and
approved the final manuscript. TLV and MAIAO confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Texas Tech University Health Sciences Center does
not require ethical approval for reporting individual cases or case
studies.
Patient consent for publication
Written informed consent was obtained from the
patient' parents for publication of anonymized data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jalanko H: Congenital nephrotic syndrome.
Pediatr Nephrol. 24:2121–2128. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bolk S, Puffenberger EG, Hudson J, Morton
DH and Chakravarti A: Elevated frequency and allelic heterogeneity
of congenital nephrotic syndrome, Finnish type, in the old order
Mennonites. Am J Hum Genet. 65:1785–1790. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Preston R, Stuart HM and Lennon R: Genetic
testing in steroid-resistant nephrotic syndrome: Why, who, when and
how? Pediatr Nephrol. 34:195–210. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fogo AB, Lusco MA, Najafian B and Alpers
CE: AJKD Atlas of Renal Pathology: Congenital nephrotic syndrome of
finnish type. Am J Kidney Dis. 66:e11–e12. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gigante M, Piemontese M, Gesualdo L,
Iolascon A and Aucella F: Molecular and genetic basis of inherited
nephrotic syndrome. Int J Nephrol. 2011(792195)2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Golob V, Nosan G, Bertok S, Frelih M,
Boštjanči E and Rus R: A novel mutation of congenital nephrotic
syndrome in a Slovenian child eventually receiving a renal
transplant. Croat Med J. 62:187–191. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Khan A, Fornes O, Stigliani A, Gheorghe M,
Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni
SR, Tan G, et al: JASPAR 2018: Update of the open-access database
of transcription factor binding profiles and its web framework.
Nucleic Acids Res. 46:D260–D266. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fornes O, Castro-Mondragon JA, Khan A, van
der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M,
Baranašić D, et al: JASPAR 2020: Update of the open-access database
of transcription factor binding profiles. Nucleic Acids Res.
48:D87–D92. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Krull M, Petrusma M, Makalowski W, Brosius
J and Schmitz J: Functional persistence of exonized mammalian-wide
interspersed repeat elements (MIRs). Genome Res. 17:1139–1145.
2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jjingo D, Conley AB, Wang J,
Mariño-Ramírez L, Lunyak VV and Jordan IK: Mammalian-wide
interspersed repeat (MIR)-derived enhancers and the regulation of
human gene expression. Mob DNA. 5(14)2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Khan MT, Ali S, Zeb MT, Kaushik AC, Malik
SI and Wei DQ: Gibbs free energy calculation of mutation in pnca
and rpsa associated with pyrazinamide resistance. Front Mol Biosci.
7(52)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Al Qahtani NH, AbdulAzeez S, Almandil NB,
Fahad Alhur N, Alsuwat HS, Al Taifi HA, Al-Ghamdi AA, Rabindran
Jermy B, Abouelhoda M, Subhani S, et al: Whole-genome sequencing
reveals exonic variation of ASIC5 gene results in recurrent
pregnancy loss. Front Med (Lausanne). 8(699672)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Majidi S, Fouts A, Pyle L, Chambers C,
Armstrong T, Wang Z, Batish SD, Klingensmith G and Steck AK: Can
biomarkers help target maturity-onset diabetes of the young genetic
testing in antibody-negative diabetes? Diabetes Technol Ther.
20:106–112. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Caridi G, Gigante M, Ravani P, Trivelli A,
Barbano G, Scolari F, Dagnino M, Murer L, Murtas C, Edefonti A, et
al: Clinical features and long-term outcome of nephrotic syndrome
associated with heterozygous NPHS1 and NPHS2 mutations. Clin J Am
Soc Nephrol. 4:1065–1072. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Golson ML and Kaestner KH: Fox
transcription factors: From development to disease. Development.
143:4558–4570. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Katoh M and Katoh M: Human FOX gene family
(Review). Int J Oncol. 25:1495–1500. 2004.PubMed/NCBI
|
|
17
|
Neznanov N, Umezawa A and Oshima RG: A
regulatory element within a coding exon modulates keratin 18 gene
expression in transgenic mice. J Biol Chem. 272:27549–27557.
1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fiszbein A, Krick KS, Begg BE and Burge
CB: Exon-mediated activation of transcription starts. Cell.
179:1551–1565.e17. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Stergachis AB, Haugen E, Shafer A, Fu W,
Vernot B, Reynolds A, Raubitschek A, Ziegler S, LeProust EM, Akey
JM, et al: Exonic transcription factor binding directs codon choice
and affects protein evolution. Science. 342:1367–1372.
2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Waldrop E, Al-Obaide MAI and Vasylyeva TL:
GANAB and PKD1 variations in a 12 years old female patient with
early onset of autosomal dominant polycystic kidney disease. Front
Genet. 10(44)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Khan AH, Lin A and Smith DJ: Discovery and
characterization of human exonic transcriptional regulatory
elements. PLoS One. 7(e46098)2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Castellanos M, Mothi N and Muñoz V:
Eukaryotic transcription factors can track and control their target
genes using DNA antennas. Nat Commun. 11(540)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fontrodona N, Aubé F, Claude JB, Polvèche
H, Lemaire S, Tranchevent LC, Modolo L, Mortreux F, Bourgeois CF
and Auboeuf D: Interplay between coding and exonic splicing
regulatory sequences. Genome Res. 29:711–722. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Joynt AT, Evans TA, Pellicore MJ,
Davis-Marcisak EF, Aksit MA, Eastman AC, Patel SU, Paul KC, Osorio
DL, Bowling AD, et al: Evaluation of both exonic and intronic
variants for effects on RNA splicing allows for accurate assessment
of the effectiveness of precision therapies. PLoS Genet.
16(e1009100)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
National Center for Biotechnology
Information: Genomic variation as it relates to human health
(ClinVar); [VCV000429811.3]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000429811.3.
Accessed October 24, 2021.
|
|
26
|
Roca X, Sachidanandam R and Krainer AR:
Intrinsic differences between authentic and cryptic 5' splice
sites. Nucleic Acids Res. 31:6321–6333. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Anna A and Monika G: Splicing mutations in
human genetic disorders: Examples, detection, and confirmation. J
Appl Genet. 59:253–268. 2018.PubMed/NCBI View Article : Google Scholar
|