Introduction
Lynch syndrome (LS) is the most common hereditary
cause of colorectal cancer (CRC), accounting for 3-5% of all CRC
cases (1). LS was previously termed
hereditary nonpolyposis CRC (HNPCC) to emphasize the absence of
colon polyps and to distinguish it from the other types of
hereditary CRC, which are characterized by the presence of polyps,
such as Adenomatous Polyposis Coli and Hamartomatous Polyposis
syndrome (1). It is estimated that
LS is possibly the most common hereditary cancer syndrome, with an
overall prevalence of 1/100-1/180 in the general population
(1). LS is associated with a high
lifetime risk of developing several types of cancer, primarily CRC
(20-70% risk with an average age of diagnosis of 44-61 years),
endometrial cancer (15-70% risk with an average age of diagnosis of
44-61 years), ovarian cancer (risk 4-12% with an average age of
diagnosis of 42.5 years), gastric cancer (risk 6-13% with an
average age of diagnosis of 56 years) and for other cancers (small
intestine, brain, skin, hepatobiliary and urinary tract, overall
risk 15%) (2). The etiology of LS
is an inherited germline mutation in one of the Mismatch Repair
(MMR) genes-MLH1 (3p22), MSH2 (2p21), MSH6 (2p16), PMS2 (7p22),
MLH3 (14q24), MSH3 (5q14), MSH5 (6p21) or MLH2 (2q32) (3). The MMR system is responsible for
repairing single base mismatches and small insertions and deletions
that occur predominantly during replication (4). According to Knudson's ‘two-hit’
hypothesis, failure of the MMR repair system is a consequence of
bi-allelic inactivation of MMR genes (classical tumor suppressor
genes) (3). Therefore, individuals
who are carriers of one germline mutation in these genes are simply
predisposed to cancer. If the somatic mutation in the second
wild-type allele occurs during the carrier's lifetime, a cancer
will develop. The somatic mutation in the corresponding wild-type
allele is typically a point mutation (3).
A deficiency in the MMR complex leads to a high
mutation rate, especially in repetitive DNA sequences (dispersed
sequence elements that make up ~3% of our genome and are usually
polymorphic), the so-called microsatellites (MS) (4) This condition is termed MS instability
(MSI) and is a specific feature of LS-associated cancers (in ~95%
of all cases) (5). Currently, there
are two methods to establish the stability of MSs. One is a
molecular test that is based on the detection of amplified MS loci
by PCR. The analysis is performed on tumor DNA (extracted from
tissue embedded in paraffin) and allows the classification of tumor
tissues as high MSI (MSI-H)-deficient mismatch repair (dMMR), or
low MSI (MSI-L)-MMR functioning properly (6). The other method is based on
immunohistochemical (IHC) detection of proteins, encoded by MMR
genes (MLH1, MSH2, MSH6 and PMS2). MMR deficiency is defined by
loss of expression of some of the four MMR proteins (6). There is no consensus on which of the
two tests is preferable for CRC as they have similar performance
characteristics in detecting LS (7-10),
while for endometrial cancer the preferred test option is IHC,
which has a sensitivity of 100% vs. 56.3% for the MSI test, with
similar specificity (11,12).
Identification of families with LS is important for
the effectiveness of surveillance strategies in affected
individuals and the prevention of cancer in their relatives. In
clinical practice, there are two major guidelines for
identification of individuals and families with LS: The Amsterdam
criteria (AC) and the Bethesda guidelines. The AC (adopted in 1990)
was used to identify families with CRC eligible for molecular
analysis of MMR deficiency (13).
Later, these criteria were updated to ACII, including other
LS-related cancers (14). It was
found that these criteria were very restrictive, resulting in
omission of ~68% of patients with LS (15). The second set of guidelines, the
Bethesda guidelines, were later developed and expanded the clinical
criteria for LS screening, taking into account the MSI status of
the tumor tissue. The Bethesda guideline panel includes five
MSs-two mononucleotide and three dinucleotide repeats (16). However, even with the updated
Bethesda criteria, a large number of patients with LS remain
underdiagnosed (17). According to
the latest recommendations of the European Hereditary Tumour Group
and the European Society of Coloproctology all colorectal and
endometrial carcinomas should be tested for MMR deficiency
(18). In cases of established MMR
deficiency, analysis of a germline mutation in the MMR genes is
recommended for precise patient therapy, to improve clinical
surveillance and to reduce cancer morbidity and mortality rates in
the families of LS patients (18).
Knowledge of the molecular mechanisms (type of mutation) and
genotype-phenotype correlations enhance the efficiency of genetic
counseling in patients with LS. In the present report, the case of
familial LS with early onset of cancer and a detected pathogenic
splice donor variant in the MSH2 gene is described.
Materials and methods
Patients
The patients (proband and his mother) were referred
to the Center of Medical Genetics at the University Hospital ‘Dr.
Georgi Stranski’ (Pleven, Bulgaria) for germline genetic testing.
Blood samples were obtained (in an EDTA plastic tube) from the
patient and his relatives (mother and uncle) after obtaining
informed consent.
IHC procedure
All tumor samples used in the present study were
collected after obtaining informed consent for participation in the
study. Tumor specimens from the proband's uncle were fixed in 10%
buffered formalin for 24-36 h at room temperature, dissected and
paraffin embedded. A pathologist selected 5 µm thick parallel
sections of representative invasive tumor material and normal
mucosa, and the tissue sample was confirmed to contain cancerous
tissue using hemoxylin and eosin staining, which was performed as
routine. Epitope retrieval time for all tumor sections was 20 min
at 97˚C in DAKO PT Link (cat. no. PT100/PT101).
Tumor sections were stained with the following
antibodies (all from Dako, Agilent Technologies, Inc., and all came
ready to use): ES05-Monoclonal mouse Anti-Human MutL Protein
Homolog 1, (cat. no. IR079), FE11-Monoclonal mouse Anti-Human MutS
Protein Homolog 2 (cat. no. IR085), EP49-Monoclonal rabbit
Anti-Human MutS Protein Homolog 6 (cat. no. IR086), and
EP51-Monoclonal rabbit Anti-Human Postmeiotic Segregation Increased
2 (cat. no. IR087) for MLH1, MSH2, MSH6 and PMS2 respectively.
Incubation time for all antibodies was 20 min at room temperature.
A Dako Agilent Autostainer Link 48 slide stainer was used according
to the manufacturer's protocol. The external negative controls were
the negative reagent controls in the kit. For internal positive
controls, normal colonic mucosa, stromal cells and stromal
lymphocytes from the same patients. Results were analyzed manually
by a pathologist. Expression was reported as: Normal, (retained
expression) nuclear expression in >10% tumor cells and retained
expression in the internal control or Negative, (loss of
expression) 0% expression in tumor cells and retained expression in
the internal control.
Germline pathogenic variant
detection
Genomic DNA was isolated from each blood sample
using a MagCore Genomic DNA Whole blood kit (MagCore®)
according to the manufacturer's protocol. The genetic testing of
the proband and his mother was performed by next generation
sequencing (NGS). The Trusight Cancer Sequencing Panel (Illumina,
Inc.) was used for library preparation according to the
manufacturer's protocol. The pan-hereditary cancer panel contained
oligo probes targeting 94 genes and 284 SNPs associated with
increased cancer predisposition. The procedures were performed
following the manufacturer's instructions. Qualified libraries were
sequenced on the Illumiina NextSeq 550 platform with a 2x150 bp
configuration (Illumina, Inc.). Reads were aligned to the reference
human genome hg19. Data output files (gVCF) were imported into
BaseSpace Variant Interpreter (Illumina, Inc.). Custom filters
(minimum read depth of 20x per variant and excluded silent
variants) were created to improve variant annotation and
interpretation. The five-tier terminology system of the American
College of Medical Genetics and Genomics was used for variant
classification (19), including:
Pathogenic (P), Likely Pathogenic (LP), Variant of Unknown clinical
significance (VUS), Likely Benign (LB) and Benign (B). The variants
automatically annotated by the software were manually checked in
the primary human genome databases: ClinVar (www.ncbi.nlm.noh.gov/clinvar), dbSNP (www.ncbi.nlm.noh.gov/projrct/SNP) and
Ensembl (http://www.ensembl.org).
The familial germline mutation in exon 8 of the MSH2
gene detected by NGS was screened in the proband's uncle using
direct Sanger sequencing. Primer pairs were designed using the
Primer blast tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)
to specifically amplify the coding exon 8 of the MSH2 gene and
exon-intron boundaries. The primer sequences were: Forward,
5'-GTGGGAAGCTTTGAGTGCTAC-3' and reverse,
5'-ATCCACTGTCCACAAAGGTGC-3'). PCR amplification of the DNA template
was performed using AmpliTaq Gold™ 360 Master Mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions. Briefly, the reaction mixture
consisted of 5 µl AmpliTaq Gold™ 360 Master Mix (2X), 3
µl PCR primers (0.8 µM each) (Applied Biosystem; Thermo Fisher
Scientific, Inc.), 1 µl DNA template (10 ng) and 1 µl
UltraPure™ DNase/RNase-Free Distilled Water (Applied
Biosystems; Thermo Fisher Scientific, Inc.). PCR amplification was
run on a GeneAmp™ PCR System 9700 (Applied Biosystems;
Thermo Fisher Scientific, Inc.) using the following thermocycling
conditions: Initial denaturation at 95˚C for 10 min; followed by 35
cycles of denaturation at 95˚C for 30 sec, annealing at 58˚C for 30
sec and extension at 72˚C for 45 sec; with a final extension step
of 72˚C for 5 min. Amplicon sequencing was performed using a
BigDye™ Terminator v.3.1 Cycle Sequencing Kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and an Applied
Biosystems™ 3130xl Genetic Analyser (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol.
Results
The proband (index patient) was a 26-year-old man,
diagnosed with metastatic CRC at the age of 25. He presented to the
surgical clinic complaining of diarrhea (4-5 times per day) for 8
months, rectal bleeding, fatigue and loss of weight and appetite.
Laboratory investigations revealed macrocytic anemia. A CT scan
detected an infiltrating rectal tumor (~10 cm) and a hypodense
liver lesion (5 mm) as well as mesenteric and iliac
lymphadenopathy. Colonoscopy revealed an ulcerative-infiltrative
tumor, occupying almost the entire circumference of the rectum.
Histopathological examination of endoscopic biopsy specimens
indicated moderately differentiated rectal adenocarcinoma. The
patient underwent the first surgery under general
anesthesia-exploratory laparotomy with deep anterior resection of
the rectum with total mesorectal excision with descending
rectostomy ‘end to end’, transverse colonoplasty,
temporary/protective ileostomy and cystofix. The patient's second
operation was the resection of the liver lesions (1.1 and 0.6 cm),
the histopathological examination of which confirmed the
preliminary suspicion of colorectal metastasis. A molecular test
for MSI was performed on the pathological specimen of the
colorectal tumor. The result showed microsatellite instability at 6
of 7 loci. The recommendation to the patient was to undergo
germline genetic analysis with sequencing of the MSH2 and MSH6
genes. The patient was referred to our Center of Medical genetics
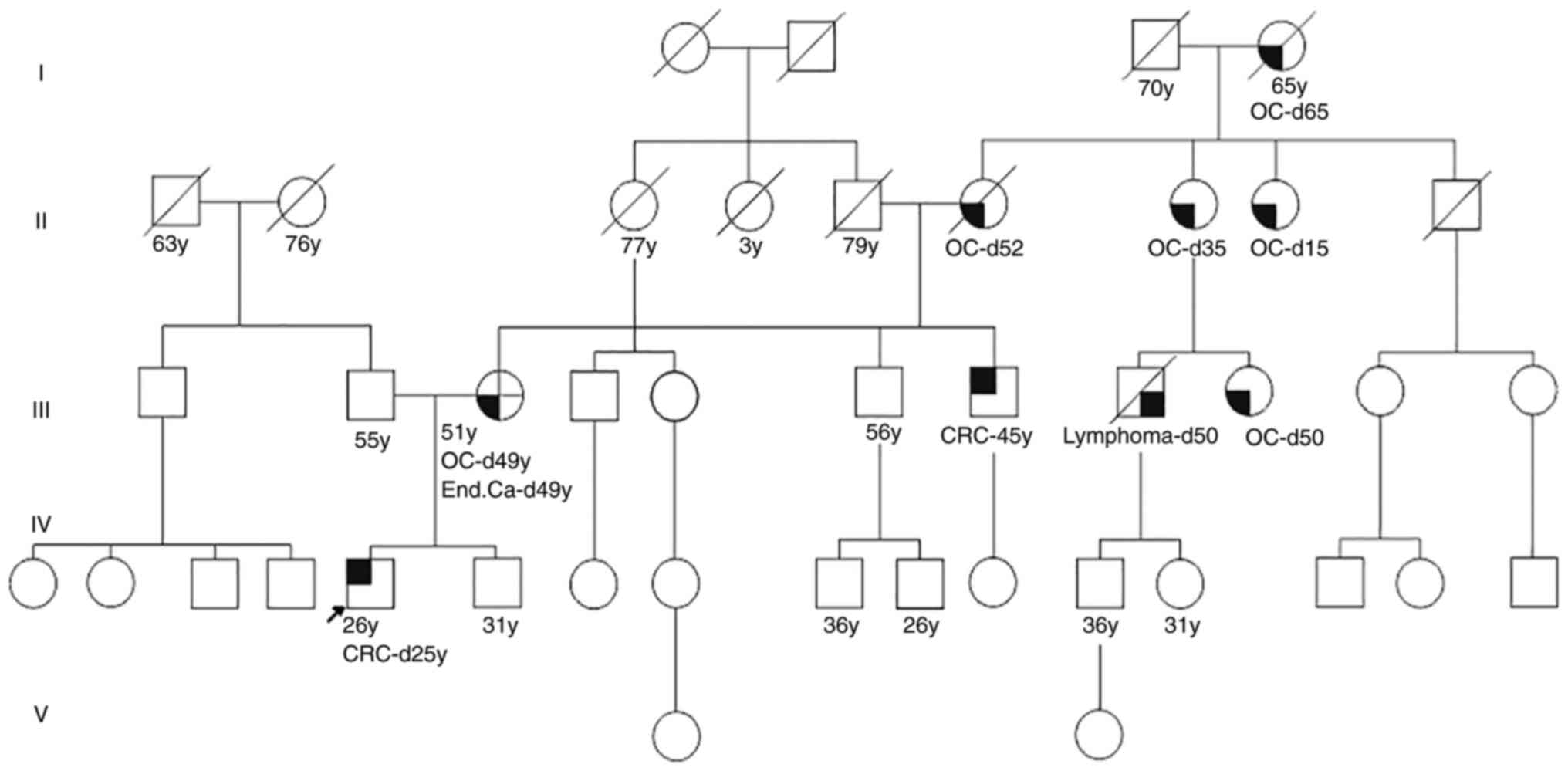
for testing. Genealogy revealed seven relatives with LS-related
cancer. The first-degree relative (mother) was diagnosed with
synchronous cancers (highly to moderately differentiated
endometrial adenocarcinoma and moderately differentiated ovarian
cystadenocarcinoma) at the age of 49 and underwent bilateral
salpingo-oophorectomy. In addition, five other relatives (II and
III degree) were diagnosed with ovarian cancer, two of them with
early onset (16 years and 35 years old; Fig. 1).
NGS of the proband and his mother detected a
pathogenic variant of MSH2, c.1386+1G >A (NM_000251.3), in both
individuals. The genetic counselor's recommendations for the
affected individuals (mother and son), in accordance with the NCCN
guideline for LS and specifically for MSH2-LS, were a high-quality
colonoscopy to be performed and repeated every 1-2 years (20). Genetic testing for the pathogenic
variant c.1386+1G >A in the MSH2 gene was recommended to the I
and II-degree relatives of the proband. One of the proband's uncles
(45 years old) was aware of his sister's genetic results and
underwent a high-quality colonoscopy, which revealed cancer in the
flexura coli-hepatica, and was the reason for a
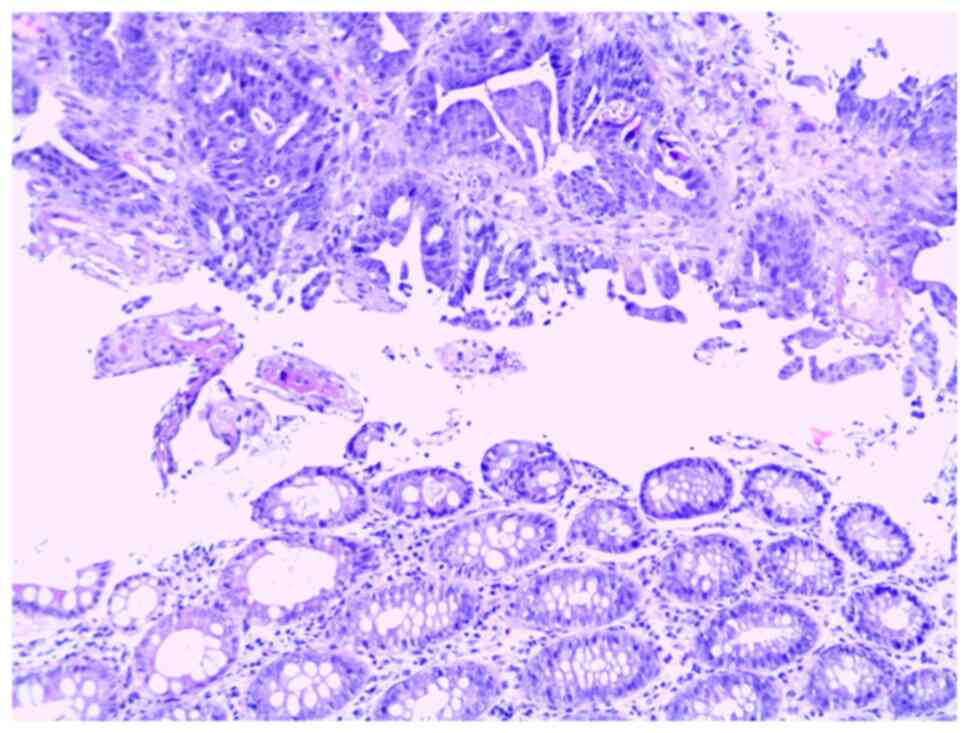
laparoscopy-assisted right colectomy. The histological result of
tumor formation in the colon showed two components, the first with
the morphology of a mucinous adenocarcinoma with extreme
extracellular mucus production and ring cells, and the second
component of a moderately to poorly differentiated adenocarcinoma
(Fig. 2). The tumor tissue was
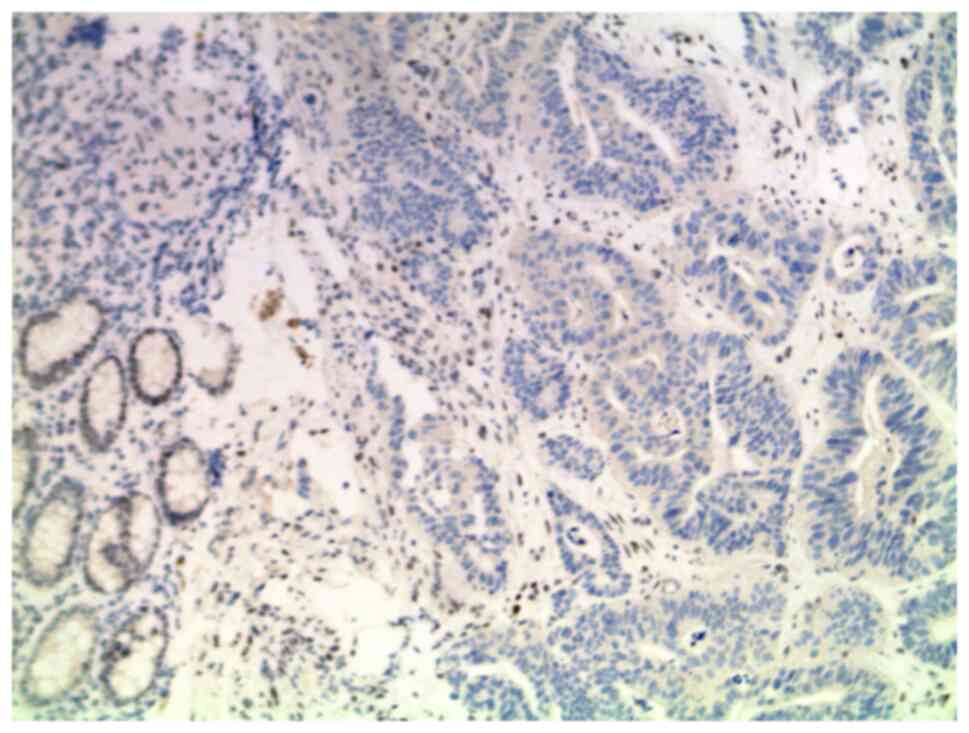
examined for MMR deficiency by IHC and the loss of MSH2 protein
expression was revealed, correlating with the carrier status of the
familial genetic variant in the MSH2 gene (Fig. 3). Subsequently, the pathogenic
variant (c.1386+1G >A) in the MSH2 gene was confirmed by Sanger
sequencing in the uncle.
Discussion
LS is one of the most common Mendelian cancer
predisposition syndromes, and the resultant cancers are typically
localized to the colon and endometrium. LS is associated with other
types of cancer locations, including the ovaries, urinary tract,
other parts of the digestive system (stomach, small intestine and
hepatobiliary tract) and the brain (1,2). The
most important criteria (according to Amsterdam criteria) for
clinical diagnosis of LS is a family history of colon cancer,
although the latest guidelines recommend all patients with colon
and endometrial cancer (regardless of their family history) are
tested for LS (18). In the present
report, the case was not a typical LS family, as the proband had no
family history of CRC, but did have a family history of ovarian and
endometrial, and this may explain the delayed diagnosis.
The second major clinical characteristic of LS is an
early age of onset of the cancer (<50 years old). In the present
case, the proband was diagnosed at the age of 25 years, and
additionally, two other III-degree relatives (with ovarian cancer)
were diagnosed at the age of 16 and 35 years old.
Tumor localization in LS-related CRC occurs with
equal frequency in the proximal colon, distal colon and the rectum
(21), consistent with the present
case. The proband presented with rectal cancer and his uncle with
right CRC. Studies have identified the morphological features of
CRC specific to LS-related tumors, and include a greater likelihood
of a poorly differentiated tumor, a medullary morphology and a
mucinous component (22,23), and these features were observed in
the case of CRC in the proband's uncle. In contrast, other studies
have shown there are no distinguishing features of LS-related CRC
(24,25), and this was the case in the proband
who presented with a moderately differentiated adenocarcinoma of
the rectum, supporting the need for a universal genetic screening
panel for LS in all patients with CRC and MSI-H or impairment of
MMR genes as assessed using IHC.
LS is caused by a germline mutation in DNA MMR
genes, primarily MLH1, MSH2, MSH6 or PMS2(3). In the present case, the pathogenic
variant c.1386+1G >A (NM_000251.3) was found in the MSH2 gene in
three of the affected family members. The MLH1 and MSH2 genes
(defined as ‘major’ MMR genes) are crucial for the DNA repair
mechanism, and the majority (71 and 84%, respectively) of LS cases
are due to germline mutations in these two genes (2,26-28).
Amongst all MMR genes, the MSH2 variant is associated with the
highest risk of different cancer localizations; although it is
typically associated with CRC, as well as endometrial and ovarian
cancer (29). In carriers of the
MSH2 variant, the cumulative cancer incidence is as follows: 46.6%
(females) and 51.4% (males) for CRC, 48.9% for endometrial cancer,
17.4% for ovarian cancer, 18.7% (women) and 17.6% (men) for
ureteral and renal cancer, and 23.8% for prostate cancer (30). The pattern of inheritance of MMR
pathogenic variants and cancer predisposition is autosomal dominant
with a 50% risk for offspring of the affected individual.
In accordance with previous literature, in the
described MSH2 familial case of LS, most of the affected members
were diagnosed with extracolonic localization of the cancer, in the
endometrium and/or ovaries (30).
The reported frequency of the pathogenic germline
MMR variants in the general population is as follows: 0.051%
(1:1,946) for MLH1 mutations, 0.035% (1:2,841) for MSH2, 0.132%
(1:758) for MSH6 and 0.140% (1:714) for PMS2(31). Amongst all MMR genes, MSH2 mutations
have the lowest frequency in the general population, but they are
the most common cause of LS due to its major role in the MMR
mechanism and the presumed highest penetrance of variants (5).
The most common type of mutation in MMR genes in
sporadic CRC and LS differs; however, this comparison between
sporadic and hereditary CRC with deficiency of MMR, was not in the
scope of the present report. In the described clinical case, the
proband had a family history of a I degree relative diagnosed with
synchronous cancer (endometrial and ovarian cancer), a clinical
feature of LS. Thus, genetic screening for germline mutations in
MMR genes was performed. The most common germline mutations in the
MSH2 gene are point mutations (nonsense, missense or alterations at
the highly conserved splice site position AG/GT) or small
insertions/deletions (frameshift) (27,32).
The pathogenic variant was found to be c.1386+1G >A
(NM_000251.3) in MSH2 (https://www.ncbi.nlm.nih.gov/clinvar/variation/90641/).
The variant is located in a canonical splice site and impairs mRNA
splicing, resulting in a significantly altered protein as a result
of exon skipping, truncation or inclusion of intronic material.
Several computational tools predict a significant impact of the
variant on normal splicing. Some publications reported experimental
evidence showing that the variant leads to exon 8 skipping
(33,34). The variant c.1386+1G >A was
absent in 248,984 healthy controls (gnomAD); however, it has also
been described in multiple individuals with HNPCC (33-35).
These data suggest that the variant is likely to be associated with
disease. In the present case, this variant was detected in three
affected relatives from the family. Taking all the data together,
the variant is classified as a pathogenic variant (19).
Diagnosis of LS in the present family was based on
the following criteria: Early age of onset of CRC in the proband,
synchronous cancer (endometrial and ovarian) in a I degree
relative, a strong family history of a LS-related cancer (ovarian),
and the detected pathogenic variant in the MSH2 gene. The other
affected family members were not available for testing, but their
early age of onset was suggestive of their carrier status. When
interpreting the test results during genetic counseling,
consideration should be given not only to the detected genetic
mutation, but also the personal and family history of the
patient.
In the family described, there was no need for
additional recommendations for the proband and his relative
II-degree relative, as they had already undergone colorectal
surgery. For the proband's mother, a high quality colonoscopy
(repeated in 1-2 years) was recommended. All unaffected family
members at risk were advised to undergo genetic testing and, in the
case of confirmation of being a carrier of the MSH2 variant, to
undergo a high-quality colonoscopy at the age of 20 years (5 years
earlier than the age at which the cancer occurred in the proband).
Female relatives at risk were advised to be alert for any abnormal
uterine bleeding or postmenopausal bleeding (uterine cancer can be
detected early by symptoms), and about prevention of ovarian cancer
(due to the late onset of clinical symptoms) in MSH2 carriers,
bilateral salpingo-oophorectomy should also be considered. Usually
the timing of risk-reducing surgery should take into account
reproductive history, menopausal status, comorbidities and family
history.
Due to the positive family history of ovarian cancer
at an early age and the familial pathogenic MSH2 variant (which is
associated with a particularly high risk of ovarian cancer),
prophylactic surgery should be performed as early as possible. In
women with incomplete reproduction, ovarian cancer prevention
should include transvaginal ultrasound screening along with a serum
test for CA-125.
The presented familial cancer case with the detected
pathogenic variant in the MSH2 gene may contribute to
genotype-phenotype correlation in LS cases. Carriers of a splice
site mutation in MSH2 with a very early age of onset of CRC were
identified in the present study. The unusual presentation in the
family-predominantly with ovarian cancer-is consistent with
previous literature showing that MSH2 mutation carriers had a
higher preponderance of extracolonic tumors. The early detection of
CRC in the uncle of the proband emphasizes the role of genetic
counseling in cases of hereditary cancer syndrome-for prevention
and effective surveillance strategies.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the European Regional
Development Fund with the leading organization Medical
University-Pleven (grant no. BG05M2OP001-1.002-0010-C01).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/submitters/507074/).
Authors' contributions
SLP recruited the patients. ZBK, SLP, KSK and KTP
collected clinical and biological data. ZBK and SEN performed the
molecular analysis. SLP was responsible for diagnosis and treatment
of patients. ZBK analyzed the data. ZBK, SLP and KSK were involved
in the writing and revision of the manuscript. SEN and KTP reviewed
and revised the manuscript. All authors have read and approved the
final manuscript. ZBK and SLP confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
This study was approved by the Ethics Commission of
the Medical University-Pleven (Pleven, Bulgaria. All the
participants in the study were >18 years of age and provided
written informed consent to participate in the study.
Patient consent for publication
Written informed consent for publication of their
data was obtained from all patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frankel W, Arends M, Frayling IM and
Nagtegaal ID: Lynch syndrome: Genetic tumour syndromes of the
digestive system. In: World Health Organization Classification of
Tumours of the Digestive System. 5th edition. IARC Press, Lyon,
2019.
|
|
2
|
Idos G and Valle L: Lynch syndrome. In:
GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA,
Wallace SE, Bean LJH, Mirzaa G and Amemiya A (eds). University of
Washington, Seattle, WA, 1993-2022.
|
|
3
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel implications in molecular diagnosis of Lynch syndrome.
Gastroenterol Res Pract. 2017(2595098)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Payseur BA, Jing P and Haasl RJ: A genomic
portrait of human microsatellite variation. Mol Biol Evol.
28:303–312. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Boland CR: Recent discoveries in the
molecular genetics of Lynch syndrome. Fam Cancer. 15:395–403.
2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chen ML, Chen JY, Hu J, Chen Q, Yu LX, Liu
BR, Qian XP and Yang M: Comparison of microsatellite status
detection methods in colorectal carcinoma. Int J Clin Exp Pathol.
11:1431–1438. 2018.PubMed/NCBI
|
|
7
|
Snowsill T, Coelho H, Huxley N,
Jones-Hughes T, Briscoe S, Frayling IM and Hyde C: Molecular
testing for Lynch syndrome in people with colorectal cancer:
Systematic reviews and economic evaluation. Health Technol Assess.
21:1–238. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Snowsill T, Huxley N, Hoyle M,
Jones-Hughes T, Coelho H, Cooper C, Frayling I and Hyde C: A
systematic review and economic evaluation of diagnostic strategies
for Lynch syndrome. Health Technol Assess. 18:1–406.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Wedden S, Miller K, Frayling IM, Thomas T,
Chefani A, Miller K, Hamblin A, Taylor JC and D'Arrigo C:
Colorectal cancer stratification in the routine clinical pathway: A
district general hospital experience. Appl Immunohistochem Mol
Morphol. 27:e54–e62. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
National Institute for Health and Care
Excellence (NICE): Molecular testing strategies for Lynch syndrome
in people with colorectal cancer: Diagnostics guidance [DG27].
NICE, London, 2020. https://www.nice.org.uk/guidance/dg27. Accessed April
24, 2020.
|
|
11
|
Ryan NAJ, McMahon R, Tobi S, Snowsill T,
Esquibel S, Wallace AJ, Bunstone S, Bowers N, Mosneag IE, Kitson
SJ, et al: The proportion of endometrial tumours associated with
Lynch syndrome (PETALS): A prospective cross-sectional study. PLoS
Med. 17(e1003263)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T,
Burn J, Cornes JM, Crawford R, Eccles D, Frayling IM, Ghaem-Maghami
S, et al: The manchester international consensus group
recommendations for the management of gynecological cancers in
Lynch syndrome. Genet Med. 21:2390–2400. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vasen HF, Mecklin JP, Watson P, Utsunomiya
J, Bertario L, Lynch P, Svendsen LB, Cristofaro G, Müller H, Khan
PM, et al: Surveillance in hereditary nonpolyposis colorectal
cancer: An international cooperative study of 165 families. The
International collaborative group on HNPCC. Dis Colon Rectum.
36:1–4. 1993.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Vasen H, Watson P, Mecklin J and Lynch H:
New clinical criteria for hereditary nonpolyposis colorectal cancer
(HNPCC, Lynch syndrome) proposed by the international collaborative
group on HNPCC. Gastroenterology. 116:1453–1456. 1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Barnetson RA, Tenesa A, Farrington SM,
Nicholl ID, Cetnarskyj R, Porteous ME, Campbell H and Dunlop MG:
Identification and survival of carriers of mutations in DNA
mismatch-repair genes in colon cancer. N Engl J Med. 354:2751–2763.
2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rodriguez-Bigas MA, Boland CR, Hamilton
SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin
L and Srivastava S: A national cancer institute workshop on
hereditary nonpolyposis colorectal cancer syndrome: Meeting
highlights and Bethesda guidelines. J Natl Cancer Inst.
89:1758–1762. 1997.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hampel H, Frankel WL, Martin E, Arnold M,
Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman
JA, et al: Feasibility of screening for Lynch syndrome among
patients with colorectal cancer. J Clin Oncol. 26:5783–5788.
2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Seppälä TT, Latchford A, Negoi I, Sampaio
Soares A, Jimenez-Rodriguez R, Sánchez-Guillén L, Evans DG, Ryan N,
Crosbie EJ, Dominguez-Valentin M, et al: European guidelines from
the EHTG and ESCP for Lynch syndrome: An updated third edition of
the Mallorca guidelines based on gene and gender. Br J Surg.
108:484–498. 2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Available on: https://www2.tri-kobe.org/nccn/guideline/colorectal/english/genetics_colon.pdf.
(In Japanese).
|
|
21
|
Leclerc J, Vermaut C and Buisine MP:
Diagnosis of lynch syndrome and strategies to distinguish
Lynch-related tumors from sporadic MSI/DMMR tumors. Cancers
(Basel). 13(467)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Young J, Simms LA, Biden KG, Wynter C,
Whitehall V, Karamatic R, George J, Goldblatt J, Walpole I, Robin
SA, et al: Features of colorectal cancers with high-level
microsatellite instability occurring in familial and sporadic
settings: Parallel pathways of tumorigenesis. Am J Pathol.
159:2107–2116. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yamada R, Yamaguchi T, Iijima T, Wakaume
R, Takao M, Koizumi K, Hishima T and Horiguchi SI: Differences in
histological features and PD-L1 expression between sporadic
microsatellite instability and Lynch-syndrome-associated disease in
Japanese patients with colorectal cancer. Int J Clin Oncol.
23:504–513. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yearsley M, Hampel H, Lehman A, Nakagawa
H, de la Chapelle A and Frankel WL: Histologic features distinguish
microsatellite-high from microsatellite-low and
microsatellite-stable colorectal carcinomas, but do not
differentiate germline mutations from methylation of the MLH1
promoter. Hum Pathol. 37:831–838. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hemminger JA, Pearlman R, Haraldsdottir S,
Knight D, Jonasson JG, Pritchard CC, Hampel H and Frankel WL:
Histology of colorectal adenocarcinoma with double somatic
mismatch-repair mutations is indistinguishable from those caused by
Lynch syndrome. Hum Pathol. 78:125–130. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yurgelun MB, Kulke MH, Fuchs CS, Allen BA,
Uno H, Hornick JL, Ukaegbu CI, Brais LK, McNamara PG, Mayer RJ, et
al: Cancer susceptibility gene mutations in individuals with
colorectal cancer. J Clin Oncol. 35:1086–1095. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Serrano D and Arteaga CE: Molecular
diagnosis of hereditary nonpolyposis colorectal cancer (Lynch
syndrome). Rev Fac Med. 64:537–542. 2016.
|
|
28
|
Pećina-Šlaus N, Kafka A, Salamon I and
Bukovac A: Mismatch repair pathway, genome stability and cancer.
Front Mol Biosci. 7(122)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li X, Liu G and Wu W: Recent advances in
Lynch syndrome. Exp Hematol Oncol. 10(37)2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dominguez-Valentin M, Sampson JR, Seppälä
TT, Ten Broeke SW, Plazzer JP, Nakken S, Engel C, Aretz S, Jenkins
MA, Sunde L, et al: Cancer risks by gene, age, and gender in 6350
carriers of pathogenic mismatch repair variants: Findings from the
Prospective Lynch syndrome database. Genet Med. 22:15–25.
2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Win AK, Jenkins MA, Dowty JG, Antoniou AC,
Lee A, Giles GG, Buchanan DD, Clendenning M, Rosty C, Ahnen DJ, et
al: Prevalence and penetrance of major genes and polygenes for
colorectal cancer. Cancer Epidemiol Biomark Prev. 26:404–412.
2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mangold E, Pagenstecher C, Friedl W,
Mathiak M, Buettner R, Engel C, Loeffler M, Holinski-Feder E,
Müller-Koch Y, Keller G, et al: Spectrum and frequencies of
mutations in MSH2 and MLH1 identified in 1,721 German families
suspected of hereditary nonpolyposis colorectal cancer. Int J
Cancer. 116:692–702. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Auclair J, Busine MP, Navarro C, Ruano E,
Montmain G, Desseigne F, Saurin JC, Lasset C, Bonadona V, Giraud S,
et al: Systematic mRNA analysis for the effect of MLH1 and MSH2
missense and silent mutations on aberrant splicing. Hum Mutat.
27:145–154. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Betz B, Theiss S, Aktas M, Konermann C,
Goecke TO, Möslein G, Schaal H and Royer-Pokora B: Comparative in
silico analyses and experimental validation of novel splice site
and missense mutations in the genes MLH1 and MSH2. J Cancer Res
Clin Oncol. 136:123–134. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Baert-Desurmont S, Coutant S, Charbonnier
F, Macquere P, Lecoquierre F, Schwartz M, Blanluet M, Vezain M,
Lanos R, Quenez O, et al: Optimization of the diagnosis of
inherited colorectal cancer using NGS and capture of exonic and
intronic sequences of panel genes. Eur J Hum Genet. 26:1597–1602.
2018.PubMed/NCBI View Article : Google Scholar
|