Introduction
β-Thalassemia and hemoglobin E (Hb E), two globin
gene defects characterized by β-globin gene mutations, lead to
reduced (β+), absent (β0), or abnormal
(βE) β-globin chain synthesis. In Thailand,
β-thalassemia and Hb E are very common, with frequencies varying
from 3-9 and 10-60%, respectively (1,2). In
Southeast Asia, particularly in Thailand, β-thalassemia/Hb E and
homozygous β-thalassemia are common β-thalassemia diseases
(3). Phenotypic variations in
disease severity have been observed in β-thalassemia disease
ranging from mild to severe clinical phenotypes (4-6).
Clinical presentation of severe cases (β-thalassemia major-like
phenotype) occurs between 6 and 24 months (7). Genetic factors affecting unbalanced
globin chain synthesis in β-thalassemia disease are primary and
secondary genetic modifiers of disease severity such as the type of
β-thalassemia mutation (primary modifier) and coinheritance of
α-thalassemia and polymorphisms associated with Hb F levels
(secondary modifiers) (8-10).
The type of β-thalassemia mutation represents
β+ or β0. In Thailand, in addition to Hb E,
the three most common β-thalassemia mutations reported are as
follows: Codons 41/42 (-TTCT), codon 17 (A>T) and IVS II-654
(C>T) (3,10). In the southern Thai population, IVS
I-5 (G>C), codon 19 (A>G), and Hb Malay were also common
after codons 41/42 (-TTCT), which are the most common in all
regions of Thailand (11-14).
Additionally, genetic variations at three major loci (HBB
cluster, HBS1L-MYB, and BCL11A) have been associated
with fetal hemoglobin levels and disease phenotypes in
β-thalassemia disease (15-17).
In Thailand, several SNPs located in the HBB cluster,
HBS1L-MYB, and BCL11A have been identified by two
genome-wide association studies with different platforms (17,18).
Several informative SNPs for predicting disease severity in Thai
and Malaysian β0-thalassemia/Hb E patients have recently
been developed (19). In Thailand,
the average life expectancy in β-thalassemia/Hb E patients is ~30
years (20,21). Several genetic and environmental
factors as well as the treatment and management of each patient
have been associated with life expectancy. Cardiovascular
complications are a common cause of death in β-thalassemia major
due to iron overload (21,22). According to the current management
of patients with safe blood transfusion and iron chelation, the
life expectancy in thalassemia major was comparable with that in
thalassemia intermedia (23).
Therefore, proper management could be considered at the age of
presentation (age at onset) of each patient to extend the life
expectancy of severe cases. This study aimed to determine the
updated spectrum of β-thalassemia mutations to predict the
contribution of genetic modifiers to disease severity, age at
onset, and predicted life expectancy in southern Thai β-thalassemia
patients.
Materials and methods
Ethical statement
Ethical clearance of the study protocol was obtained
from the Institutional Review Board of Walailak University (Nakhon
Si Thammarat, Thailand; approval no. 12/030). Written informed
consent was obtained from all patients/guardians. All experiments
were performed in accordance with relevant guidelines and
regulations.
Study population
A cross-sectional study was conducted on
β-thalassemia patients enrolled from thalassemia clinics, pediatric
departments (child patients), and internal medicine departments
(adult patients) from 6 different provinces between July 2012 and
August 2014. All patients were diagnosed with β-thalassemia/Hb E
(Hb types of EF or EFA) or homozygous β-thalassemia (Hb types of
A2F or A2FA) based on the clinical
manifestations, a complete blood count, and hemoglobin analysis.
DNA analyses were then performed for confirmation of
β-thalassemia/Hb E and homozygous β-thalassemia. Disease severity
was classified using a scoring system according to 6 independent
parameters as follows: the hemoglobin level at steady state, the
age at first blood transfusion, a requirement for blood
transfusion, the spleen size (or splenectomy status), the age at
disease presentation and growth development (24).
Hematological analysis
A complete blood count was performed using the
Sysmex XN-1000 automated hematology analyzer (Sysmex Corporation).
Hemoglobin analysis was performed using an automated
high-performance liquid chromatography (HPLC-Variant II
β-thalassemia short program, Bio-Rad Laboratories, Inc.).
DNA extraction and measurement of the
concentration and purity of the extracted genomic DNA
Genomic DNA was extracted from peripheral blood
leukocytes using the Genomic DNA Extraction Kit (Geneaid) according
to the manufacturer's instructions. The concentration and purity of
gDNA were measured at wavelengths of 260 and 280 nm using a
Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.).
Characterization of globin gene
mutations
β-Thalassemia mutations were characterized using
polymerase chain reaction (PCR)-based methods. Common β-globin gene
mutations were first identified by PCR-reverse dot blot
hybridization (PCR-RDB) (25), all
probe sequences are listed in Table
SI. or a multiplex amplification refractory mutation system
(MARMS) (26) followed by multiplex
gap-PCR (deletion type) (14). The
Hb E allele was confirmed by real-time PCR-high resolution melting
(HRM) analysis as described previously (27). Mutational characterization of DNA
samples with negative results from PCR-RDB or MARMS-PCR and other
PCR-based methods were further identified by automated DNA
sequencing (Solgent Co., Ltd) of the whole HBB gene as
described previously (28), the
additional forward and reverse primer sequences were
5'-CGGCTGTCATCACTTAGACC-3' and 5'-GCAGCTTGTCACAGTGCAG-3',
respectively (product size, 598 bp). Common α-globin gene
deletions, including α-thalassemia 1 alleles (--SEA and
--THAI) and α-thalassemia 2 alleles (-α3.7
and -α4.2), were characterized by multiplex gap-PCR
whereas Hb constant spring and Hb Pakse alleles were identified by
allele-specific PCR as described in previous studies (29,30).
All primer sequences are shown in Table SII.
Single nucleotide polymorphism (SNP)
genotyping
Four SNPs [rs7482144 (XmnI), rs2071348
(HBBP1) rs766432 and rs9376074] from three representative
regions (HBB cluster, BCL11A and HBS1L) were
selected for genotyping. PCR-restriction fragment length
polymorphism (RFLP) was used to characterize the genotypes of
rs7482144, rs2071348 and rs766432 as described previously and the
primer sequences for SNP genotyping were as follows: rs7482144
forward primer, 5'-GGCCTAAAACCACAGAGAGT-3' and reverse primer,
5'-CCAGAAGCGAGTGTGTGGAA-3'; rs2071348 forward primer,
5'-GGCACCTTTGCTACACTGAG-3' and reverse primer,
5'-TCATCATTCGGAGGGAAACA-3', and rs766432 forward primer,
5'-AAAATCTCAGAATACAAAGGGC-3' and reverse primer,
5'-GTTAGGGAAGGGGATTGAC-3' (27,31,32).
Additionally, SNP genotyping of rs9376074 was performed using
PCR-HRM and the primer sequences were: Forward,
5'-GAAGATGAAGCTAAGGTTTGG-3' and reverse, 5'-TCTGACTCCTCAAATGCC-3'
(27).
Statistical analysis
Descriptive statistics were used to describe the
spectrum of β-globin gene mutations, disease severity
score/grouping, and hematological parameters of the patients.
Clinical and hematological data from different severities of
patients were compared using a χ2 test for categorical
variables and the Kruskal-Wallis test for continuous variables
(non-normally distributed data) between the mild, moderate and
severe groups using SPSS (version 26.0. IBM Corp.). Single SNP
association analyses for disease severity, age at onset, and
predicted life expectancy were performed in the recessive and
allelic models using a Pearson's χ2 test and/or Fisher's
exact test. The P-value, odds ratios (OR), and 95% conference
intervals (CIs) were calculated to compare genotype and allele
frequencies using 2x2 contingency tables in publicly accessible
statistical software (http://vassarstats.net/odds2x2.html). P<0.05 was
considered to indicate a statistically significant difference. The
clustered bar and the 100% stacked column were constructed using
Infogram (https://infogram.com/) and Microsoft
Excel, respectively.
Results
Patient classification according to α-
and β-globin genotypes and disease phenotypes
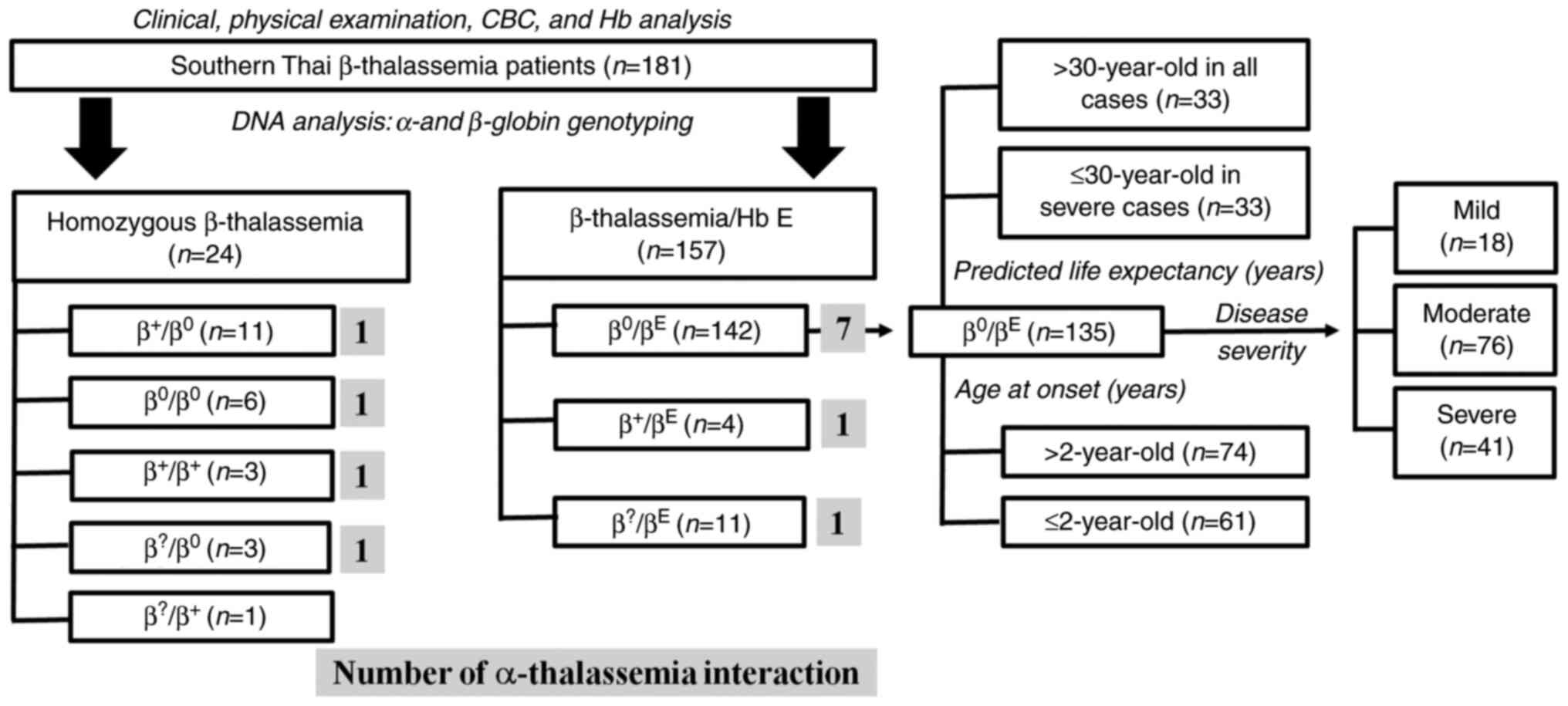
A total of 181 β-thalassemia patients were enrolled
and classified according to their β-globin genotypes, including 24
homozygous β-thalassemia and 157 β-thalassemia/Hb E patients.
Clinical data, physical examination, complete blood count, and
hemoglobin analysis were used for evaluating disease phenotypes. In
addition, α-thalassemia interactions were found in 4 homozygous
β-thalassemia and 9 β-thalassemia/Hb E patients. A total of 135
β0-thalassemia/Hb E without α-thalassemia interactions
were divided into 3 categories according to their predicted life
expectancy, disease severity, and the age of onset. According to an
average life expectancy of β0-thalassemia/Hb E patients
(30 years of age), the 135 β0-thalassemia/Hb E patients
were divided into two groups according to age: ≤30 (33 patients
from severe cases who were predicted to have a lower life
expectancy) and >30 (33 patients from all cases who were
predicted to have a higher life expectancy). The second category
was grouped according to disease severity, including 18 mild cases,
76 moderate cases, and 41 severe cases. The third category was
grouped according to age at onset, including 61 cases with an age
at onset ≤2 years old and 74 cases with an age at onset >2 years
old (a threshold of 2 years of age was selected as this is the
cutoff point between thalassemia major and thalassemia intermedia),
as shown in Fig. 1.
Disease severity and primary and
secondary genetic modifiers in southern Thai β-thalassemia
patients
The 181 patients with β-thalassemia were classified
as 34 mild cases, 95 moderate cases, and 52 severe cases and
further subdivided into 6 groups according to the β-globin
genotypes (Table I). Among the 181
patients with β-thalassemia, β0-thalassemia/Hb E
accounted for 78% and was grouped into 21 mild cases, 80 moderate
cases, and 41 severe cases. All β-thalassemia patients with
β+/β+ and β+/βE
genotypes were grouped as the mild disease phenotype and
demonstrated that the primary modifier, the type of β-globin
mutation, can predict disease severity. Additionally, the effect of
the secondary genetic modifier, α-thalassemia interaction, was
demonstrated as β-thalassemia patients who carry α-thalassemia 2
(-α3.7/αα); 50% had mildly affected and 50% had
moderately affected phenotypes. Moreover, one patient with
β-thalassemia who carried Hb CS heterozygote had a mildly affected
phenotype. In contrast, homozygous β0-thalassemia
patients were mostly scored as a severely affected phenotype. A
homozygous β0-thalassemia patient coinheritance with
α-thalassemia 2 heterozygote had a mildly affected phenotype.
Rarely did a patient with compound heterozygosity for IVSII-837,
T>G (unclear β+ or β0), and Hb E have a
moderate disease phenotype. Among 14 patients (8%), only one
β-thalassemia mutation could be identified leaving 14
uncharacterized β-thalassemia alleles.
 | Table IPrimary (β-thalassemia mutations) and
secondary modifiers (α-thalassemia mutations) of disease severity
in the southern Thai β-thalassemia cohort. |
Table I
Primary (β-thalassemia mutations) and
secondary modifiers (α-thalassemia mutations) of disease severity
in the southern Thai β-thalassemia cohort.
| | Disease severity
(score range) | |
|---|
| β-globin gene
genotype | Mild (0.0-3.5) | Moderate
(4.0-7.0) | Severe
(7.5-10.0) | Total, n |
|---|
|
β+/β+ or
β+/βE | | | | |
|
nt -28,
A>G/nt -28, A>G | 2 | 0 | 0 | 2 |
|
nt -28,
A>G/Codon 19, A>G | 1 [1]b | 0 | 0 | 1 |
|
nt -28,
A>G/Codon 26, G>A (Hb E) | 4 [1]a | 0 | 0 | 4 |
|
Total, n
(%) | 7 (100%) | 0 (0%) | 0 (0%) | 7 |
| β0 (or
β+severe form)/β+ | | | | |
|
3.5-kb
HBB deletion/Codon 19, A>G | 0 | 1 | 0 | 1 |
|
Codons
41/42, -TTCT/Codon 19, A>G | 1 [1]a | 2 | 2 | 5 |
|
Codon 17,
A>T/Codon 19, A>G | 0 | 0 | 2 | 2 |
|
IVS I-1,
G>T/Codon 19, A>G | 0 | 0 | 1 | 1 |
|
IVS II-654,
C>T/Codon 19, A>G | 0 | 1 | 1 | 2 |
|
Total, n
(%) | 1 (9%) | 4 (36%) | 6 (55%) | 11 |
| β0 (or
β+severe form)/βE | | | | |
|
Codons 8/9,
+G/Hb E | 0 | 2 | 0 | 2 |
|
Codon 17,
A>T/Hb E | 1 | 14 | 8 | 23 |
|
IVS I-1,
G>T/Hb E | 4 | 5 | 2 | 11 |
|
IVS I-5,
G>C/Hb E | 5 [1]a | 26 [1]a | 15 | 46 |
|
Codon 35,
C>A/Hb E | 0 | 1 | 1 | 2 |
|
Codon 41
(-C), TTC>TT-/Hb E | 1 [1]a | 2 | 1 | 4 |
|
Codons
41/42, -TTCT/Hb E | 5 | 22 [3]a | 12 | 39 |
|
Codon 43,
G>T/Hb E | 0 | 1 | 0 | 1 |
|
Codons
71/72, +A/Hb E | 1 [1]a | 0 | 0 | 1 |
|
IVS II-654,
C>T/Hb E | 1 | 4 | 2 | 7 |
|
105-bp
HBB deletion/Hb E | 0 | 2 | 0 | 2 |
|
3.5-kb
HBB deletion/Hb E | 3 | 1 | 0 | 4 |
|
Total, n
(%) | 21 (15%) | 80 (56%) | 41 (29%) | 142 |
| β0 (or
β+severe form)/β0 | | | | |
|
Codon 17,
A>T/Codon 17, A>T | 0 | 0 | 1 | 1 |
|
Codon 17,
A>T/IVS I-1, G>T | 0 | 0 | 1 | 1 |
|
Codon 17,
A>T/Codons 41/42, -TTCT | 0 | 0 | 1 | 1 |
|
Codons
41/42, -TTCT/Codons 41/42, -TTCT | 0 | 1 | 1 | 2 |
|
IVS I-5,
G>C/3.5-kb HBB deletion | 1 [1]a | 0 | 0 | 1 |
|
Total, n
(%) | 1 (17%) | 1 (17%) | 4 (66%) | 6 |
| β+ or
0/βE (rare type, unclear β+ or
β0) | | | | |
|
IVS II-837,
T>G/Hb E | 0 | 1 | 0 | 1 |
|
Total, n
(%) | 0 (0%) | 1 (100%) | 0 (0%) | 1 |
|
βUnch/βE,
βUnch/β0 and
βUnch/β+ | | | | |
|
Uncharacterized
mutation/Hb E | 3 | 7 [1]a | 0 | 10 |
|
Uncharacterized
mutation/105 bp HBB deletion | 0 | 1 [1]a | 0 | 1 |
|
Uncharacterized
mutation/IVS I-5, G>C | 0 | 1 | 0 | 1 |
|
Uncharacterized
mutation/Codon 15, -T | 0 | 0 | 1 | 1 |
|
Uncharacterized
mutation/Codon 19, A>G | 1 | 0 | 0 | 1 |
|
Total, n
(%) | 4 (29%) | 9 (64%) | 1 (7%) | 14 |
|
β+-Thalassemia (2 alleles), n
(%) | 7 (100%) | 0 (0%) | 0 (0%) | 7 |
| α-Thalassemia
interaction/Hb CS, n (%) | 7 (54%) | 6 (46%) | 0 (0%) | 13 |
| Total β-thalassemia
patients, n (%) | 34 (19%) | 95 (52%) | 52 (29%) | 181 |
Clinical and hematological
characteristics of southern Thai β0-thalassemia/Hb E
patients in different age groups
Several patient characteristics were significantly
different between the 2 age groups (≤30 years old and >30 years
old), such as age, age at presentation, age at first blood
transfusion, frequency of blood transfusion, spleen status, and
growth development (P<0.05). Approximately 97% of the
≤30-year-old group required regular blood transfusion. A greater
number of splenectomized patients was highly observed in the
≤30-year age group. According to the standard Thai growth chart,
the >30-year-old group was found mostly (>73%) in the
10th-25th percentile, whereas in the ≤30 years age group it was
mostly observed (>67%) in the 3rd-10th percentile (Table II). Obvious differences in all
clinical and hematological findings between the 3 disease severity
groups were observed (Table
SIII).
| Table IIBaseline, clinical, and hematological
profiles of 66 southern Thai β0-thalassemia/Hb E
patients without α-thalassemia interactions categorized by age
groups. |
Table II
Baseline, clinical, and hematological
profiles of 66 southern Thai β0-thalassemia/Hb E
patients without α-thalassemia interactions categorized by age
groups.
| | Age group | |
|---|
| Patient
characteristics | ≤30-year-old,
severe cases, n=33 | >30-year-old,
all cases, n=33 | P-value |
|---|
| Sex, n (%) | | | 0.210d |
|
Male | 11(33) | 16(48) | |
|
Female | 22(67) | 17(52) | |
| Age (years), mean ±
SD | 14.1±4.73 | 46.2±12.63 |
<0.0001c,e |
| Baseline Hb (g/dl),
mean ± SD | 6.7±0.99 | 6.9±1.32 | 0.705e |
| Age at presentation
(years), mean ± SD | 1.4±0.93 | 18.6±18.15 |
<0.0001c,e |
| Age at first
transfusion (years), mean ± SD | 1.8±1.48 | 22.8±19.94 |
<0.0001c,e |
| Requirement for
regular blood transfusion, n (%) | 32(97) | 19(58) | 0.0001c,d |
| Spleen size (cm),
mean ± SD | 7.2±4.80 | 7.1±5.52 | 0.966e |
| Splenectomy, n
(%) | 23(70) | 12(36) | 0.007a,d |
| Growth development:
Height, n (%) | | | 0.0003b,d |
|
≤P3-10 | 23(70) | 8(25) | |
|
≥P10-25 | 10(30) | 24(75) | |
| Growth development:
Weight, n (%) | | | 0.0013a,d |
|
≤P3-10 | 22(67) | 9(27) | |
|
≥P10-25 | 11(33) | 24(73) | |
An updated β-thalassemia mutational
spectrum in southern Thai β-thalassemia patients
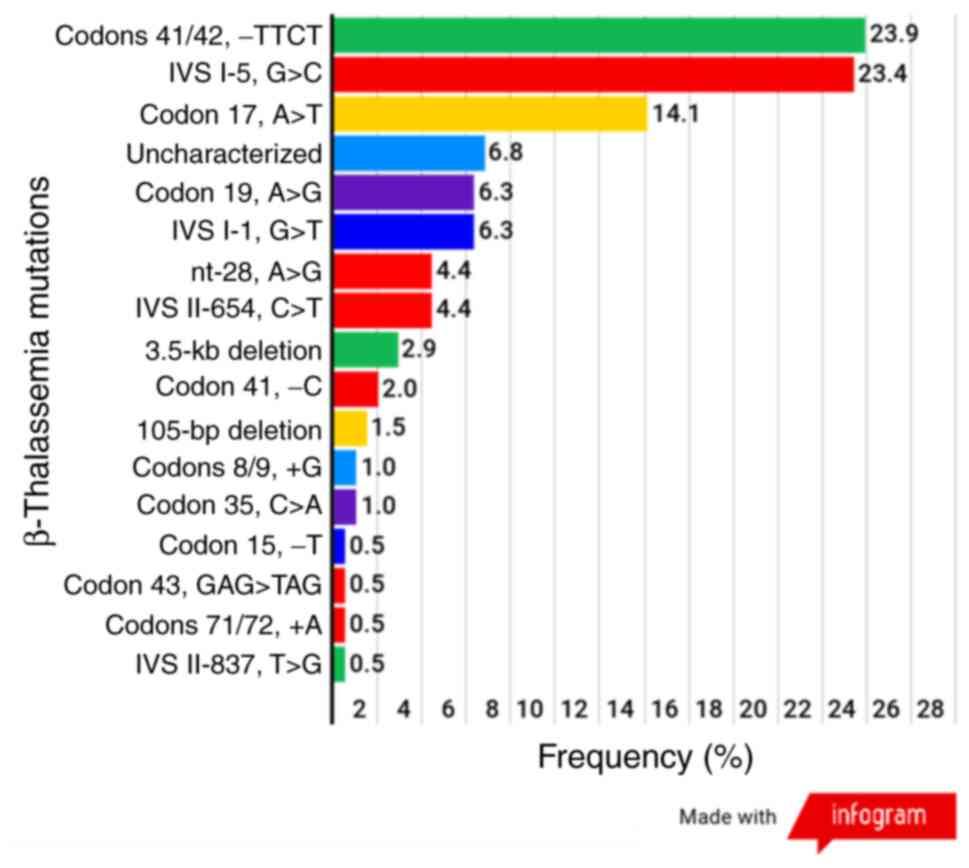
In the present study, 181 patients with
β-thalassemia including, 24 with homozygous β-thalassemia and 157
with β-thalassemia/Hb E disease, were recruited. In total, 362
β-globin alleles from 181 β-thalassemia patients and 16 different
mutations were identified, among which 3 common mutations accounted
for 61.4% (Hb E was not included) as follows: Codons 41/42, -TTCT;
IVS I-5, G>C and codon 17, A>T with frequencies of 23.9,
23.4, and 14.1%, respectively. All 3 of the most common mutations
were categorized as β0 (codons 41/42, -TTCT and codon
17, A>T) or the severe form of β+ (IVS I-5, G>C).
A total of 14 alleles of the β-globin gene, from 14 β-thalassemia
patients were not successfully characterized in either allele of
the β-globin gene, and these patients were grouped as having
uncharacterized β-globin gene mutations (Fig. 2).
Associations between SNPs and disease
severity and age at onset
The associations between the 4 SNPs and the disease
severity of β0-thalassemia/Hb E patients using mild and
severe disease severity groups. The XmnI polymorphism showed
a strong association with the disease severity (P=0.004; OR, 3.20;
95% CI, 1.42-7.22) (Table SIV). To
predict the age at onset of southern Thai
β0-thalassemia/Hb E patients according to the SNP
genotypes from 3 independent regions, the CC genotype of
XmnI (rs7482144) was a strong predictor and showed a
significantly increased risk for younger age at onset (P=0.004; OR,
3.13; 95% CI, 1.40-7.00). In contrast, there was no association in
BCL11A (rs766432) and HBS1L-MYB (rs9376074)
regions (Table SV). Among the 3
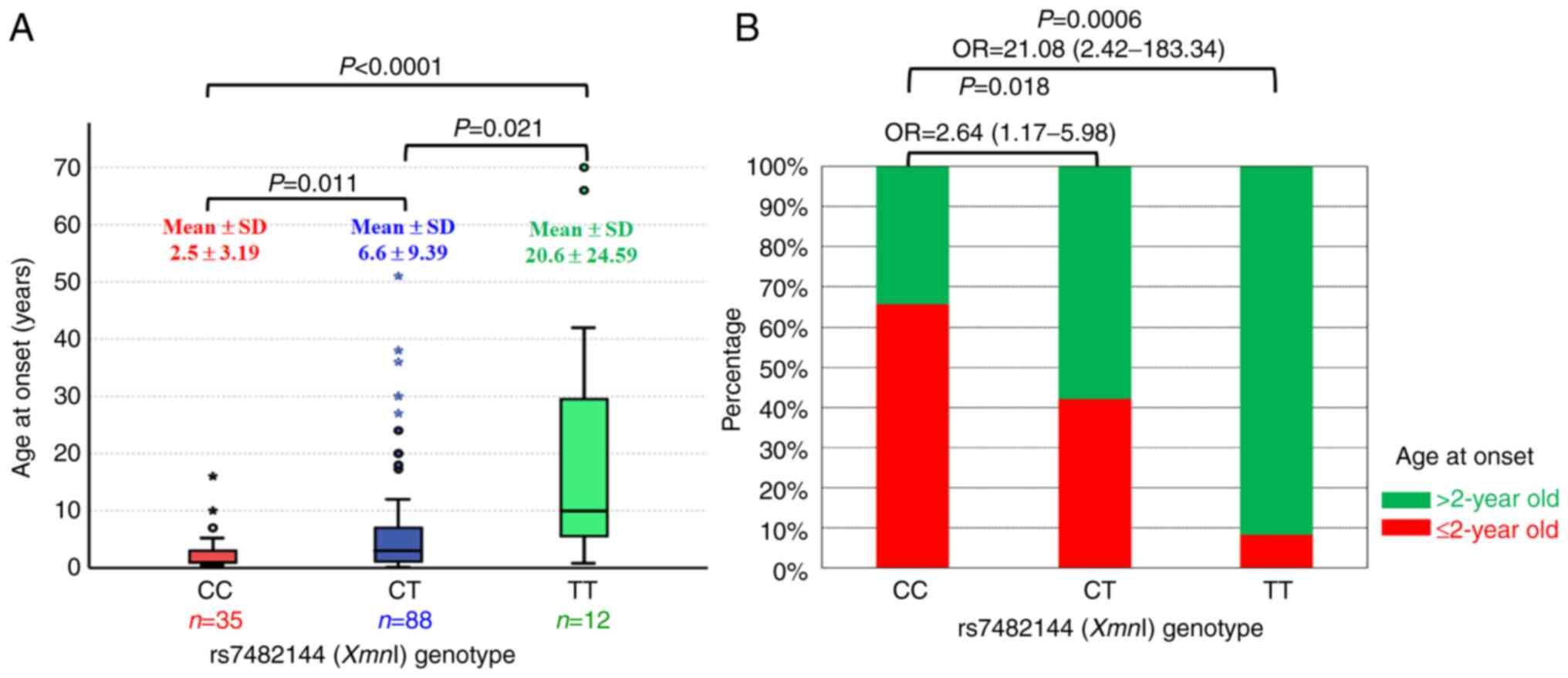
genotypes of XmnI, the mean and standard deviation of age at
onset (2.5±3.19, 6.6±9.39, and 20.6±24.59 years) were increased
according to the number of T alleles (CC, CT, and TT,
respectively). In addition, the comparisons of the mean age at
onset from each genotype were significant (P<0.05) (Fig. 3A). To apply the XmnI
genotypes for predicting the age at onset, the TT genotype was
observed in >90% of individuals in the >2 years of age group,
and the CC genotype was observed in >60% of the individuals in
the ≤2 years of age group. The frequency of the TT genotype in the
>2 years of age group was higher than that in the ≤2 years of
age group (P=0.0006; OR, 21.08; 95% CI, 2.42-183.34) (Fig. 3B).
Associations between SNPs and the
predicted life expectancy
The associations between the 4 candidate SNPs from 3
independent regions and the predicted life expectancy of
β0-thalassemia/Hb E patients were next assessed. The
XmnI (rs7482144) polymorphism showed a strong association
with the predicted life expectancy (P=0.004; OR, 6.50; 95% CI,
1.64-25.80). The CT or TT genotype of XmnI was associated
with a higher predicted lifespan than those with the CC genotype.
In addition, rs2071348 also exhibited an association with the
predicted life expectancy (P=0.016). In contrast, rs766432 and
rs9376074 demonstrated no association with the predicted life
expectancy (P=0.458 and 0.438, respectively) (Table III).
| Table IIIAssociation of the 4 SNPs in 3
independent regions with the predicted life expectancy in southern
Thai β0-thalassemia/Hb E patients without α-thalassemia
interactions. |
Table III
Association of the 4 SNPs in 3
independent regions with the predicted life expectancy in southern
Thai β0-thalassemia/Hb E patients without α-thalassemia
interactions.
| | Age Range
Status | |
|---|
| SNP info |
Genotype/allele | ≤30-year-old,
severe cases, n=33 | >30-year-old,
all cases, n=33 |
P-valueb | Odds ratio (95%
Confidence interval) | Risk
Genotype/Allelea |
|---|
| rs7482144 (C/T),
HBG2 | | | | | | |
|
Genotype | CC | 13 (0.394) | 3 (0.091) | 0.004 | 6.50
(1.64-25.80) | CC |
| | CT+TT | 20 (0.606) | 30 (0.909) | | | |
|
Allele | C | 46 (0.697) | 29 (0.440) | 0.003 | 2.93
(1.43-6.00) | C |
| | T | 20 (0.303) | 37 (0.560) | | | |
| rs2071348 (A/C),
HBBP1 | | | | | | |
|
Genotype | AA | 11 (0.333) | 3 (0.091) | 0.016 | 5.00
(1.24-20.08) | AA |
| | AC+CC | 22 (0.667) | 30 (0.909) | | | |
|
Allele | A | 43 (0.652) | 28 (0.424) | 0.009 | 2.54
(1.26-5.13) | A |
| | C | 23 (0.348) | 38 (0.576) | | | |
| rs766432 (C/A),
BCL11A | | | | | | |
|
Genotype | AA | 20 (0.606) | 17 (0.515) | 0.458 | 1.45
(0.54-3.84) | AA |
| | AC+CC | 13 (0.394) | 16 (0.485) | | | |
|
Allele | A | 53 (0.803) | 49 (0.742) | 0.406 | 1.41
(0.62-3.21) | A |
| | C | 13 (0.197) | 17 (0.258) | | | |
| rs9376074 (T/C),
HBS1L | | | | | | |
|
Genotype | TT | 13 (0.394) | 10 (0.303) | 0.438 | 1.50
(0.54-4.14) | TT |
| | TC+CC | 20 (0.606) | 23 (0.697) | | | |
|
Allele | T | 42 (0.636) | 37 (0.561) | 0.374 | 1.37
(0.68-2.76) | T |
| | C | 24 (0.364) | 29 (0.439) | | | |
Cascade genetic testing of
β0-thalassemia/Hb E patients for phenotype
predictions
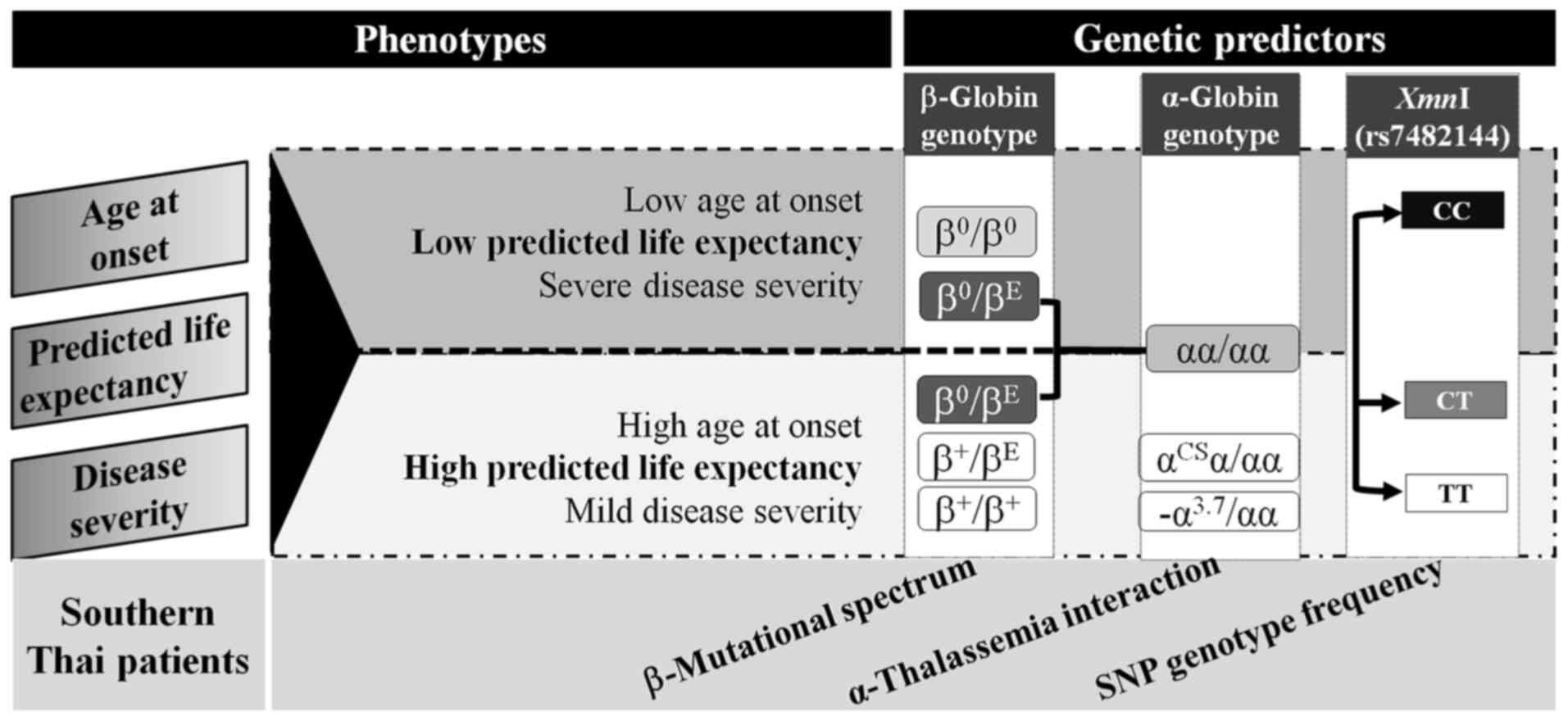
According to the overall results, the age at onset,
predicted life expectancy, and disease severity were assigned as
phenotypic variations in β-thalassemia patients. Phenotypic
variations were then classified into 2 groups: Low or high
predicted life expectancy. The genotyping of β-thalassemia
mutations, α-thalassemia interactions, and XmnI genotypes
were sequentially recommended for phenotype prediction; for
example, the CT or TT genotype of XmnI was observed in 90.9%
of β0-thalassemia/Hb E patients with high predicted life
expectancy (Fig. 4).
Discussion
β-thalassemia and Hb E are very common in Thailand,
in which the frequency of the β-thalassemia trait varies from 3 to
9%, and the frequency of Hb E is 13% on average and varies from
region to region. The frequency of Hb E is very high at the
junction of Thailand, Laos, and Cambodia at 50-60% (33). In Thailand, the number of patients
with compound heterozygotes for β-thalassemia and Hb E is higher
than that for homozygous β-thalassemia because the frequency of Hb
E is much higher than that for β-thalassemia (3,20,34).
β-thalassemia/Hb E disease showed diverse disease phenotypes
ranging from mild to severely affected patients (6,21). The
variation in disease severity in β-thalassemia patients could be
explained by β-thalassemia mutations (10), α-thalassemia interactions (35,36),
and genetic determinants of Hb F production (15,17,18,37,38),
and other factors related to the pathophysiology of β-thalassemia
(39-41).
Several genetic modifiers associated with disease severity and
fetal hemoglobin levels in β-thalassemia/Hb E patients have been
well studied in the Thai population (10,17,18).
Factors affecting life expectancy in β-thalassemia patients were a
subset of disease severity-associated genetic factors and proper
treatments such as safe blood transfusion, iron chelation, and
other supportive therapies can decrease disease complications.
However, there is no report of SNP frequency data and some rare
β-thalassemia mutations in southern Thai
β0-thalassemia/Hb E patients. According to different
genetic backgrounds and migration, the mutational spectrum of
β-thalassemia and SNP frequency in southern Thai differed in other
parts of Thailand. This phenomenon has also been observed in
various countries such as India (42-44),
Malaysia, China (45,46), and other countries (47). Therefore, the predictive performance
of the β-thalassemia mutations and SNPs would differ in each region
in the same country.
According to the primary modifier, the present study
demonstrated that all β+/β+ and
β+/βE patients were scored as mildly affected
due to the primary modifier (10,17).
The frequency of disease severity among β0/βE
southern Thai patients with mild, moderate, and severe disease
phenotypes was distributed in a different pattern than in previous
studies because of the different genetic backgrounds (17,24) of
the studied patients. This study revealed that the β-thalassemia
mutations are very heterogeneous, with a wider distribution in
southern Thailand than in other parts of Thailand. The 3 most
common β-thalassemia mutations in this study were 61.4%, which
differed from the central (72.4%), northern (83.1%), and
northeastern (80.3%) regions of Thailand (12). A total of 17 β-thalassemia
mutations, including Hb E, were identified in 181 southern Thai
β-thalassemia patients in the present study. Comparing these
results to previously published reports in the southern Thai
population, the 4 most common mutations, codons 41/42 (-TTCT), IVS
I-5 (G>C), codon 17 (A>T), and codon 19 (A>G) accounted
for 67.7% of mutations in the present study and revealed slightly
different patterns and frequencies due to the differences in the
collected sample backgrounds, such as ethnicity (11), thalassemia status (trait or disease)
(48), different provinces
(12-14)
of southern Thailand and migration (47) (Fig.
S1). Although this study recruited patients from several
provinces of southern Thailand, a similar pattern of the most
common β-thalassemia mutations was observed. The origin of patients
may explain the difference in distribution; for example, codons
41/42 (-TTCT) are very common in individuals of Chinese origin
(45,46), whereas IVS I-5 (G>C) is very
common in the Malay (49) and Asian
Indian (50) populations.
Interestingly, the present study demonstrated comparable
frequencies of codons 41/42; -TTCT (23.9%) and IVS I-5; G>C
(23.4%) because of the higher sample size of Thai-Muslim patients.
The spectrum and frequency of β-thalassemia mutations in the
southern Thai population were different from those in other regions
of Thailand (3,51). Hb Malay was found at the highest
frequency (11.7%) in the southern region of Thailand compared with
other parts of Thailand (12,13).
The frequency of Hb Malay in our study was 6.3%, ranking as the
fourth most common β-thalassemia mutation in southern Thailand.
Heterozygous β-thalassemia (IVS II-837; T>G) was first described
in Asian Indians with unclear β+ or β0
thalassemia showing a typical asymptomatic carrier, and the
incidence of this mutation was found in the Gaud Saraswat (44), Brahmins in Goa, and Karnataka
(52) states of southern India.
Phenotypes of the homozygous state of IVS II-837 (T>G) were
transfusion-dependent (52).
Interestingly, compound heterozygotes of IVS II-837 (T>G) and Hb
E were found for the first time in the present study and showed a
moderately affected phenotype with regular blood transfusion.
According to the disease phenotype from this study and previous
reports, IVS II-837 (T>G) could be categorized as
β+-thalassemia (severe form) (44,52).
Coinheritance of α-thalassemia in β-thalassemia
patients is one of the ameliorating factors due to more balanced
globin chain synthesis (35,36).
Heterozygous α-thalassemia 2 and Hb CS were found only in 7 mild
and 6 moderate cases in the present study. The α-thalassemia 1
allele was not detected in our β-thalassemia patients. A possible
reason is that the coinheritance of α-thalassemia 1 leads to mild
β-thalassemia; thus, these patients were not found in a
hospital-based sample collection (35). Furthermore, several genetic markers
in the HBB cluster (10,15,17,51),
BCL11A (15,17,19),
and HBS1L-MYB (15,17)
have been associated with fetal hemoglobin and disease severity in
several populations. In addition, mutations in human Krüppel-like
factor 1 (KLF1) were found to be associated with increased
fetal hemoglobin (Hb F) and hemoglobin A2 (Hb
A2) (53,54). KLF1 mutations have been
studied in patients with β-thalassemia/Hb E, and a higher Hb F
level was observed in the cases with KLF1 mutations
(38,51).
According to hospital-based sample collection, the
present study failed to enroll sufficient mild cases (n=18) for SNP
analysis in disease severity because the mild case has a lower
frequency of going to the hospital. However, XmnI and
rs2071348 were associated with disease severity in
β0-thalassemia/Hb E (with low power). The T allele
frequency of XmnI in mild cases (0.611) was significantly
higher than that in severe cases (0.329). No associations were
found in rs766432 and rs9376074 because of the low sample size in
mild cases (Table SIV). An
increased sample size could improve the statistical power in all
SNPs due to the similar trend of the allele frequencies (17).
Currently, the life expectancy between thalassemia
major and thalassemia intermedia is comparable due to the use of
safe blood transfusions, effective iron chelation, and improved
management of cardiovascular complications (23,55).
Proper patient management should be initiated during the age at
onset for improved quality of life and increased life expectancy.
Therefore, the prediction of the age at onset is important not only
for patient management in newborns but also for genetic counseling
in prenatal diagnosis (PND). Our study showed the association
between SNPs and the age at onset and the predicted life expectancy
of southern Thai β0-thalassemia/Hb E patients.
Interestingly, the XmnI polymorphism and rs2071348 were
associated with the age at onset and the predicted life expectancy.
The XmnI polymorphism is the strongest marker for predicting
the age at onset and the predicted life expectancy in southern Thai
patients with β0-thalassemia/Hb E. This polymorphism is
well identified in association with fetal hemoglobin levels and
disease phenotypes in different groups of populations (10,15,17-19,51).
Due to the improved and individualized management of the patients,
an improved life expectancy and quality of life were observed in
β-thalassemia patients. In addition, the life expectancy of
thalassemia major patients was similar to that of thalassemia
intermedia patients (23,55,56).
Therefore, the genetic prediction of age at onset and life
expectancy is suggested for better patient management after newborn
screening. Concerning precision medicine, the β-thalassemia
mutations and XmnI (rs7482144) polymorphism could be
simultaneously genotyped to improve genetic counseling in PND.
However, this suggested guideline should be validated on a national
scale and with considerably larger sample sizes in the future.
In summary, genetic heterogeneity and a broad
spectrum of β-globin gene mutations were observed in southern Thai
β-thalassemia patients. This study provides an updated spectrum of
β-thalassemia mutations. Hb Malay, IVS I-5 (G>C), 105-bp
deletion, and 3.5-kb deletion were primarily found in the southern
Thai population, accounting for 34.1% of all mutations. The type of
β-globin gene mutation and co-inheritance of α-thalassemia are
strong predictors of disease severity. The XmnI polymorphism
and rs2071348 were associated with the age at onset and predicted
life expectancy. However, SNPs on BCL11A and intergenic
HBS1L-MYB are required to confirm the genetic association in
a larger sample size. This study demonstrates that the XmnI
polymorphism is the best genetic predictor for age at onset and
life expectancy. Therefore, genetic prediction for age at onset and
life expectancy may be beneficial and practical during PND or
newborn screening for better genetic counseling and optimal
management.
Supplementary Material
Comparison of the frequencies (%) of
β-thalassemia mutations in southern Thailand.
List of amino-linked oligonucleotide
probes for PCR-reverse dot blot hybridization with common and rare
β-thalassemia mutations (25).
List of primer sequences of PCR-RDB,
MARMS-PCR, multiplex-gap PCR, PCR-HRM, and DNA sequencing.
Clinical and hematological findings of
the 135 southern Thai β0-thalassemia/Hb E patients
without α-thalassemia interactions.
Association of 4 SNPs in 3 independent
regions with disease severity in southern Thai
β0-thalassemia/Hb E patients without α-thalassemia
interactions.
Association of 4 SNPs in 3 independent
regions with the age at onset in 135 southern Thai
β0-thalassemia/Hb E patients without α-thalassemia
interactions.
Acknowledgements
We would like to thank Mrs. Dararat Horpet for the
technical support and all of the patients who participated in this
research project.
Funding
Funding: This study was funded by a grant from the Thailand
Research Fund (grant no. MRG5580069).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
MN designed and performed the experiments, analyzed
the data, and wrote the manuscript. PR performed PCR-HRM. TB, AC,
KS, NS, KL, and OT provided clinical data and performed the
physical examination and helped in obtaining blood specimens. SS
and SF provided DNA controls and helped to design the experiments.
All authors have read and approved the final manuscript. MN, TB,
AC, KS, NS, KL, and OT confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The study was conducted according to the
Declaration of Helsinki guidelines and approved by the Human
Research Ethics Committee of Walailak University (Nakhon Si
Thammarat, Thailand; approval no. 12/030).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fucharoen S, Winichagoon P, Siritanaratkul
N, Chowthaworn J and Pootrakul P: α- and β-thalassemia in Thailand.
Ann N Y Acad Sci. 850:412–414. 1998.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fucharoen S and Winichagoon P:
Hemoglobinopathies in Southeast Asia: Molecular biology and
clinical medicine. Hemoglobin. 21:299–319. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Thein SL, Winichagoon P, Hesketh C, Best
S, Fucharoen S, Wasi P and Weatherall DJ: The molecular basis of
β-thalassemia in Thailand: Application to prenatal diagnosis. Am J
Hum Genet. 47:369–375. 1990.PubMed/NCBI
|
|
4
|
Winichagoon P, Thonglairoam V, Fucharoen
S, Wilairat P, Fukumaki Y and Wasi P: Severity differences in
β-thalassaemia/haemoglobin E syndromes: Implication of genetic
factors. Br J Haematol. 83:633–639. 1993.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fucharoen S, Winichagoon P, Pootrakul P,
Piankijagum A and Wasi P: Variable severity of Southeast Asian
β0-thalassemia/Hb E disease. Birth Defects Orig Artic
Ser. 23:241–248. 1987.PubMed/NCBI
|
|
6
|
Fucharoen S, Ketvichit P, Pootrakul P,
Siritanaratkul N, Piankijagum A and Wasi P: Clinical manifestation
of β-thalassemia/hemoglobin E disease. J Pediatr Hematol Oncol.
22:552–557. 2000.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Galanello R and Origa R: β-thalassemia.
Orphanet J Rare Dis. 5(11)2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nuntakarn L, Fucharoen S, Fucharoen G,
Sanchaisuriya K, Jetsrisuparb A and Wiangnon S: Molecular,
hematological and clinical aspects of thalassemia major and
thalassemia intermedia associated with Hb E-β-thalassemia in
Northeast Thailand. Blood Cells Mol Dis. 42:32–35. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yamsri S, Singha K, Prajantasen T,
Taweenan W, Fucharoen G, Sanchaisuriya K and Fucharoen S: A large
cohort of β(+)-thalassemia in Thailand: Molecular,
hematological and diagnostic considerations. Blood Cells Mol Dis.
54:164–169. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Winichagoon P, Fucharoen S, Chen P and
Wasi P: Genetic factors affecting clinical severity in
β-thalassemia syndromes. J Pediatr Hematol Oncol. 22:573–580.
2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Laosombat V, Nopparatana C,
Wongchanchailert M and Wiriyasateinkul A: Molecular basis of
β-thalassemia in Thai Muslim patients in the the south of Thailand.
Southeast Asian J Trop Med Public Health. 28 (Suppl 3):S104–S105.
1997.PubMed/NCBI
|
|
12
|
Laosombat V, Fucharoen SP, Panich V,
Fucharoen G, Wongchanchailert M, Sriroongrueng W, Nopparatana C,
Kenpitak K, Maipang M and Fukumaki Y: Molecular basis of
β-thalassemia in the South of Thailand. Am J Hematol. 41:194–198.
1992.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Laosombat V, Wongchanchailert M,
Sattayesevana B and Nopparatana C: Clinical, hematological and
molecular features in Thais with β-Malay/β-thalassemia and
β-Malay/HbE. Southeast Asian J Trop Med Public Health. 28 (Suppl
3):S106–S109. 1997.PubMed/NCBI
|
|
14
|
Nopparatana C, Panich V, Saechan V,
Sriroongrueng V, Nopparatana C, Rungjeadpha J, Pornpatkul M,
Laosombat V and Fukumaki Y: The spectrum of β-thalassemia mutations
in Southern Thailand. Southeast Asian J Trop Med Public Health. 26
(Suppl 1):S229–S234. 1995.PubMed/NCBI
|
|
15
|
Lettre G, Sankaran VG, Bezerra MA, Araújo
AS, Uda M, Sanna S, Cao A, Schlessinger D, Costa FF, Hirschhorn JN
and Orkin SH: DNA polymorphisms at the BCL11A, HBS1L-MYB, and
β-globin loci associate with fetal hemoglobin levels and pain
crises in sickle cell disease. Proc Natl Acad Sci USA.
105:11869–11874. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ma Q, Abel K, Sripichai O, Whitacre J,
Angkachatchai V, Makarasara W, Winichagoon P, Fucharoen S, Braun A
and Farrer LA: β-globin gene cluster polymorphisms are strongly
associated with severity of HbE/β(0)-thalassemia. Clin
Genet. 72:497–505. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nuinoon M, Makarasara W, Mushiroda T,
Setianingsih I, Wahidiyat PA, Sripichai O, Kumasaka N, Takahashi A,
Svasti S, Munkongdee T, et al: A genome-wide association identified
the common genetic variants influence disease severity in
β0-thalassemia/hemoglobin E. Hum Genet. 127:303–314.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sherva R, Sripichai O, Abel K, Ma Q,
Whitacre J, Angkachatchai V, Makarasara W, Winichagoon P, Svasti S,
Fucharoen S, et al: Genetic modifiers of Hb
E/β0-thalassemia identified by a two-stage genome-wide
association study. BMC Med Genet. 11(51)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Munkongdee T, Tongsima S, Ngamphiw C,
Wangkumhang P, Peerapittayamongkol C, Hashim HB, Fucharoen S and
Svasti S: Predictive SNPs for β0-thalassemia/HbE disease
severity. Sci Rep. 11(10352)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fucharoen S and Weatherall DJ: The
hemoglobin E thalassemias. Cold Spring Harb Perspect Med.
2(a011734)2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fucharoen S and Winichagoon P: Clinical
and hematologic aspects of hemoglobin E β-thalassemia. Curr Opin
Hematol. 7:106–112. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cunningham MJ, Macklin EA, Neufeld EJ and
Cohen AR: Thalassemia Clinical Research Network. Complications of
β-thalassemia major in North America. Blood. 104:34–39.
2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vitrano A, Calvaruso G, Lai E, Colletta G,
Quota A, Gerardi C, Concetta Rigoli L, Pitrolo L, Cuccia L,
Gagliardotto F, et al: The era of comparable life expectancy
between τhalassaemia major and intermedia: Is it time to revisit
the major-intermedia dichotomy? Br J Haematol. 176:124–130.
2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sripichai O, Makarasara W, Munkongdee T,
Kumkhaek C, Nuchprayoon I, Chuansumrit A, Chuncharunee S,
Chantrakoon N, Boonmongkol P, Winichagoon P and Fucharoen S: A
scoring system for the classification of β-thalassemia/Hb E disease
severity. Am J Hematol. 83:482–484. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Winichagoon P, Saechan V, Sripanich R,
Nopparatana C, Kanokpongsakdi S, Maggio A and Fucharoen S: Prenatal
diagnosis of β-thalassaemia by reverse dot-blot hybridization.
Prenat Diagn. 19:428–435. 1999.PubMed/NCBI
|
|
26
|
Mirasena S, Shimbhu D, Sanguansermsri M
and Sanguansermsri T: Detection of β-thalassemia mutations using a
multiplex amplification refractory mutation system assay.
Hemoglobin. 32:403–409. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kesornsit A, Jeenduang N, Horpet D,
Plyduang T and Nuinoon M: Quantitative trait loci influencing Hb F
Levels in Southern Thai Hb E (HBB: c.79G>A) Heterozygotes.
Hemoglobin. 42:23–29. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nuinoon M, Jeenduang N, Kesornsit A,
Horpet D and Plyduang T: Hematological and molecular
characterization of a novel Hb A2 variant with
homozygous α-thalassemia-2 in a Southern Thai Individual.
Hemoglobin. 41:213–215. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chong SS, Boehm CD, Higgs DR and Cutting
GR: Single-tube multiplex-PCR screen for common deletional
determinants of α-thalassemia. Blood. 95:360–362. 2000.PubMed/NCBI
|
|
30
|
Fucharoen S, Sanchaisuriya K, Fucharoen G,
Panyasai S, Devenish R and Luy L: Interaction of hemoglobin E and
several forms of α-thalassemia in Cambodian families.
Haematologica. 88:1092–1098. 2003.PubMed/NCBI
|
|
31
|
Fucharoen S, Shimizu K and Fukumaki Y: A
novel C-T transition within the distal CCAAT motif of the
Gγ-globin gene in the Japanese HPFH: Implication of
factor binding in elevated fetal globin expression. Nucleic Acids
Res. 18:5245–5253. 1990.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Roy P, Bhattacharya G, Mandal A, Dasgupta
UB, Banerjee D, Chandra S and Das M: Influence of BCL11A,
HBS1L-MYB, HBBP1 single nucleotide polymorphisms and the HBG2 XmnI
polymorphism On Hb F levels. Hemoglobin. 36:592–599.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Fucharoen S and Winichagoon P:
Haemoglobinopathies in Southeast Asia. Indian J Med Res.
134:498–506. 2011.PubMed/NCBI
|
|
34
|
Yamsri S, Sanchaisuriya K, Fucharoen G,
Sae-Ung N, Ratanasiri T and Fucharoen S: Prevention of severe
thalassemia in Northeast Thailand: 16 years of experience at a
single university center. Prenat Diagn. 30:540–546. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Winichagoon P, Fucharoen S, Weatherall D
and Wasi P: Concomitant inheritance of α-thalassemia in
β0-thalassemia/Hb E disease. Am J Hematol. 20:217–222.
1985.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sripichai O, Munkongdee T, Kumkhaek C,
Svasti S, Winichagoon P and Fucharoen S: Coinheritance of the
different copy numbers of α-globin gene modifies severity of
β-thalassemia/Hb E disease. Ann Hematol. 87:375–379.
2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jomoui W, Tepakhan W, Yamsri S, Srivorakun
H, Fucharoen G and Fucharoen S: A novel SNP rs11759328 on Rho
GTPase-activating protein 18 gene is associated with the expression
of Hb F in hemoglobin E-related disorders. Ann Hematol. 99:23–29.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Khamphikham P, Sripichai O, Munkongdee T,
Fucharoen S, Tongsima S and Smith DR: Genetic variation of
Krüppel-like factor 1 (KLF1) and fetal hemoglobin (HbF) levels in
β0-thalassemia/HbE disease. Int J Hematol. 107:297–310.
2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Azman NF, Abdullah WZ, Hanafi S, Diana R,
Bahar R, Johan MF, Zilfalil BA and Hassan R: Genetic polymorphisms
of HbE/β-thalassemia related to clinical presentation: Implications
for clinical diversity. Ann Hematol. 99:729–735. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zarghamian P, Azarkeivan A, Arabkhazaeli
A, Mardani A and Shahabi M: Hepcidin gene polymorphisms and iron
overload in β-thalassemia major patients refractory to iron
chelating therapy. BMC Med Genet. 21(75)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Torres FF, Bernardo VS, Silva DGH, Okumura
JV and Bonini-Domingos CR: Association of FOXO3 polymorphism
(rs3800231) and clinical subphenotypes of β-thalassemic
individuals. Hematol Transfus Cell Ther: Nov 22, 2020 (Epub ahead
of print).
|
|
42
|
Kumar R, Kaur A and Agarwal S: Influence
of Xmn 1(G)γ (HBG2 c.-211 C → T) globin gene
polymorphism on phenotype of Thalassemia patients of North India.
Indian J Hematol Blood Transfus. 30:286–290. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Bandyopadhyay S, Roychowdhury K, Chandra
S, Das M and Dasgupta UB: Variable severity of β-thalassemia
patients of Eastern India: Effect of α-thalassemia and XmnI
polymorphism. Clin Exp Med. 1:155–159. 2001.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Colah RB and Gorakshakar A: Control of
thalassemia in India. Thalass Rep. 4(1955)2014.
|
|
45
|
Zhuang J, Zhang N, Wang Y, Zhang H, Zheng
Y, Jiang Y, Xie Y and Chen D: Molecular characterization analysis
of thalassemia and hemoglobinopathy in Quanzhou, Southeast China: A
large-scale retrospective study. Front Genet.
12(727233)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yin A, Li B, Luo M, Xu L, Wu L, Zhang L,
Ma Y, Chen T, Gao S, Liang J, et al: The prevalence and molecular
spectrum of α- and β-globin gene mutations in 14,332 families of
Guangdong Province, China. PLoS One. 9(e89855)2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kattamis A, Forni GL, Aydinok Y and
Viprakasit V: Changing patterns in the epidemiology of
β-thalassemia. Eur J Haematol. 105:692–703. 2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Nopparatana C, Nopparatana C, Saechan V,
Karnchanaopas S and Srewaradachpisal K: Prenatal diagnosis of α-
and β-thalassemias in southern Thailand. Int J Hematol.
111:284–292. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Abdullah UYH, Ibrahim HM, Mahmud NB,
Salleh MZ, The LK, Noorizhab MNFB, Zilfalil BA, Jassim HM, Wilairat
P and Fucharoen S: Genotype-phenotype correlation of β-thalassemia
in Malaysian population: Toward effective genetic counseling.
Hemoglobin. 44:184–189. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Sinha S, Black ML, Agarwal S, Colah R, Das
R, Ryan K, Bellgard M and Bittles AH: Profiling β-thalassaemia
mutations in India at state and regional levels: Implications for
genetic education, screening and counselling programmes. Hugo J.
3:51–62. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yamsri S, Pakdee N, Fucharoen G,
Sanchaisuriya K and Fucharoen S: Molecular Understanding of
Non-Transfusion-Dependent Thalassemia Associated with hemoglobin
E-β-Thalassemia in Northeast Thailand. Acta Haematol. 136:233–239.
2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bashyam MD, Chaudhary AK and Bhat V: The
IVS-II-837 (T>G) Appears to be a Relatively Common ‘Rare’
β-Globin Gene Mutation in β-Thalassemia patients in Karnataka
State, South India. Hemoglobin. 36:497–503. 2012.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Liu D, Zhang X, Yu L, Cai R, Ma X, Zheng
C, Zhou Y, Liu Q, Wei X, Lin L, et al: KLF1 mutations are
relatively more common in a thalassemia endemic region and
ameliorate the severity of β-thalassemia. Blood. 124:803–811.
2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Perseu L, Satta S, Moi P, Demartis FR,
Manunza L, Sollaino MC, Barella S, Cao A and Galanello R: KLF1 gene
mutations cause borderline HbA2. Blood. 118:4454–4458.
2011.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Borgna-Pignatti C, Rugolotto S, De Stefano
P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL,
Gamberini MR, Ghilardi R, et al: Survival and complications in
patients with thalassemia major treated with transfusion and
deferoxamine. Haematologica. 89:1187–1193. 2004.PubMed/NCBI
|
|
56
|
Taher AT, Bou-Fakhredin R, Kattamis A,
Viprakasit V and Cappellini MD: Improving outcomes and quality of
life for patients with transfusion-dependent β-thalassemia:
Recommendations for best clinical practice and the use of novel
treatment strategies. Expert Rev Hematol. 14:897–909.
2021.PubMed/NCBI View Article : Google Scholar
|