Introduction
Fabry disease (FD) is a rare familial sex X-linked
disorder attributed to GLA mutations. The disease is a
progressive severe genetic condition that worsens over time and is
characterized by various symptoms (1,2).
Genetic and Rare Diseases Information Center (GARD) reported 76
symptoms that patients with this disease may have (rarediseases.info.nih.gov). The FD symptoms may
develop during childhood (classic type) or middle adulthood
(atypical type); males tend to have more severe symptoms (1). The GLA mutations can cause
total or partial decreased activity of α-galactosidase A (α-Gal A)
and accumulation of glycosphingolipids, globotriaosylceramide
(Gb3/GL-3), and globotriaosylsphingosine (lyso-Gb3) in various
cells and organs including the skin, eyes, kidneys, heart, brain,
and peripheral nervous system (3,4). The
therapeutic approach for FD is enzyme replacement therapy (ERT);
this treatment is used to substitute the missing or an altered
partially functional α-Gal A (5-8).
Additionally, pharmacological chaperone (PC)
1-deoxygalactonojirimycin is used for the treatment of amenable
α-Gal A missense mutations (9-11).
However, ERT and PC cannot treat all FD symptoms and may cause
adverse side effects; thus, persistent symptoms in patients reduce
their quality of life (12).
Consequently, it is an open question whether FD clinical
manifestations are solely the result of α-Gal A malfunction. The
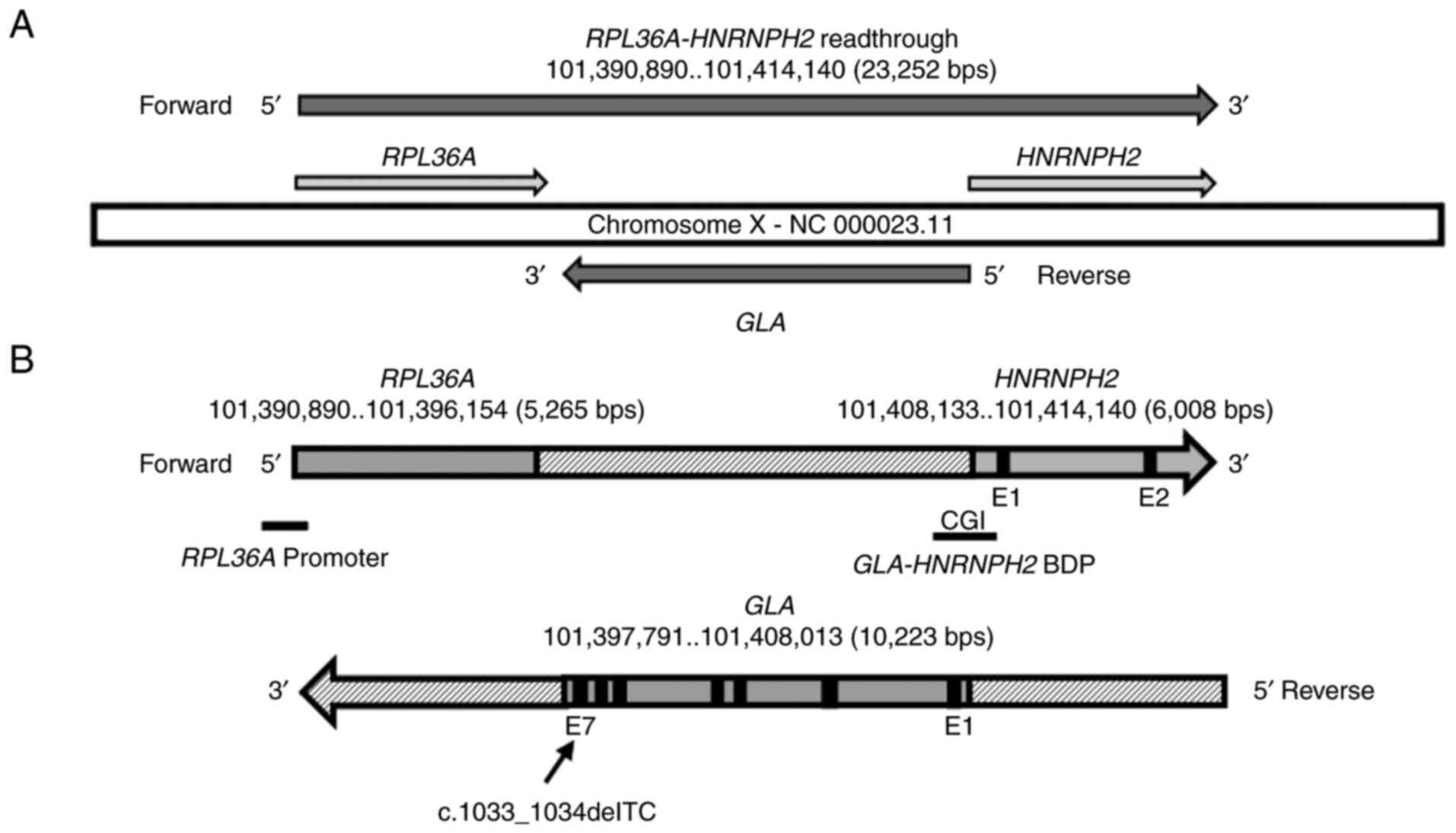
GLA gene is located at chrX: 101397791-101408013, mapped at
the reverse strand of the RPL36A-HNRNPH2 readthrough locus
(chrX: 101390890-101414140) that appears on the forward strand of
the complete genomic region NC_000023.11. The RPL36A-HNRNPH2
readthrough locus is composed of RPL36A and HNRNPH2
genes mapped at chrX: 101390890-101396154 and chrX:
101408133-101414140, respectively. Ensembl (https://useast.ensembl.org/index.html) and
ClinVar-NCBI (https://www.ncbi.nlm.nih.gov/clinvar/) databases
showed that GLA, HNRNPH2, and RPL36A genes are mapped
in the RPL36A-HNRNPH2 readthrough genomic region in humans
and are likely involved in FD and other genetic conditions
(Table SI).
In our prior research (12), human cells (adult epidermal
keratinocytes, renal glomerular endothelial cells, renal epithelial
cells, and 293 T cells) were used to show the function of the
bidirectional promoter (BDP) in the expression of GLA and
HNRNPH2 loci, which are paired in a divergent manner. One of
the primary BDP features is the presence of a susceptible CpG
Island (CGI) to DNA methylation (12). The promoters' methylation is
associated with various diseases, and the level of CGI methylation
is a further factor in the severity of the disease (12-14).
In the current study, peripheral blood from FD patients from the
same family carrying a GLA deletion mutation was used to
investigate the possible accumulative effects of GLA
deletion mutation with BDP methylation levels on the severity of
the disease in FD patients and on the GLA and HNRNPH2
expression. Blood was chosen to avoid the use of more invasive
tissues, such as skin or kidney biopsy.
Materials and methods
Participants
The control group included four healthy individuals,
two females (C1 and C2) and two males (C3 and C4). The FD group
included two females (FD1 and FD2) and two males (FD3 and FD4) from
the same family (Table I). The
healthy and FD patients' participants age ranges were 28-45 and
18-39 years respectively. The mean age of the healthy and FD
participants was 34.3±7.5 and 31.5±9.3 years respectively. The
inclusion criteria for participants were written informed consent,
collection of venous blood, understanding and agreeing to comply
with the planned study procedures, and for FD patients they had to
be part of the family being studied and had to be diagnosed with
FD. The females could not be or trying to get pregnant during the
duration of the study. Exclusion criteria included being unable or
unwilling to provide consent to participate in this study. The
healthy subjects, two males and two females were recruited for the
study from the outpatient clinic of the Texas Tech University
Health Sciences Center (Amarillo). The healthy subjects were
referred by primary care physicians and were not on any medications
and had no significant medical problems. The healthy and FD
participants agreed to the use of their samples and data in
scientific research. The blood samples were collected between May
2016-May 2018. The severity of FD in the four patients was measured
using the Mainz Severity Score Index (MSSI) and the Fabry Outcome
Survey adaptation of the Mainz Severity Score Index (FOS-MSSI)
(15).
 | Table IList of the FD patients and their
clinicopathological criteria. |
Table I
List of the FD patients and their
clinicopathological criteria.
| | Clinicopathological
criteria |
|---|
| Patients | Sex/age, years | Symptoms | Family history | ERT | RRT | Notes |
|---|
| FD1 | F/35 |
Arrhythmia/Bradycardia, Peripheral
Neuropathy | Yes | Yes | No | ERT stopped |
| FD2 | F/34 |
Arrhythmia/Bradycardia, Peripheral
Neuropathy | Yes | Yes | No | ERT stopped |
| FD3 | M/18 | Multiple
angiokeratomas, Peripheral Neuropathy | Yes | Yes | No | Died of an
unrelated cause |
| FD4 | M/39 | Progressive loss of
kidney function, ESRD Hypertension, Depression, stroke,
angiokeratomas | Yes | Yes | Yes | Died of a from
stroke |
Genomics databases
The sequences of the GLA, HNRNPH2, and
RPL36A and the predicted sequence of the BDP for the
divergently paired genes GLA and HNRNPH2 were
searched in NCBI-Gene (https://www.ncbi.nlm.nih.gov/gene/), UCSC genome
browser (https://genome.ucsc.edu/), and the
Ensembl genomics databases (https://useast.ensembl.org/index.html). Genomic tools
in the databases were used to retrieve the sequences and identify
the forward and reverse strands. The BLAT tool (https://genome.ucsc.edu/cgi-bin/hgBlat?command=start)
was used to verify map position and the EMBOSS Programs (EMBL-EBI)
(https://www.ebi.ac.uk/Tools/emboss/)
were used, EMBOSS cpgplot to discover CpG Islands (https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)
and EMBOSS matcher (https://www.ebi.ac.uk/Tools/psa/emboss_matcher/) and
EMBOSS needle (https://www.ebi.ac.uk/Tools/psa/emboss_needle/) to
perform DNA alignment analysis.
RNA and DNA extraction from blood and
RT-qPCR
The methods used were based on our previous study
(12). Briefly, an RNA/DNA
purification kit (cat. no. 48700; Norgen Biotek Corp.), was used to
extract RNA and DNA from blood cells. RT-qPCR was performed in
triplicate using a Taq Universal SYBR Green One-Step Kit (Bio-Rad
Laboratories, Inc.) and quantified using a Bio-Rad iCylcer iQ
system and the included software (Bio-Rad Laboratories, Inc.). The
Bio-Rad thermocycling protocol was optimized per experimental
requirements using the designed primers for GLA and
HNRNPH2 and the reference gene HPRT1 (12). IDT tool (idtdna.com)
was used to design the specific primers for GLA and
HNRNPH2 expression. The designed primer sets were:
HNRNPH2 forward, 5'-AGTAGTTCTGGTCGTCGTCTA-3' and reverse,
5'-ACACACCAACCTCTAACGATAC-3'; and GLA forward,
5'-AGGTTACCCGCGGAAATTTAT-3' and reverse, 5'-GAAACGAGGGCCAGGAAG-3'.
Normalization and analysis for the target genes were performed
using HPRT1 as a reference gene (forward primer,
5'-TGAGGATTTGGAAAGGGTGT-3' and reverse,
5'-GAGCACACAGAGGGCTACAA-3'). Relative expression measurements were
performed according to the 2-∆∆Cq method (16,17).
Methylation status of CGI
The CGI (323 bp) methylation of GLA-HNRNPH2
BDP was tested using bisulfite DNA treatment and MSP analysis as
described by Al-Obaide et al (12). Purified genomic DNA (100 ng) from
blood was treated using the Methylamp DNA Modification kit
(Epigentek Group Inc.), and the converted DNA was cleaned,
captured, and eluted using R6 (Modified DNA Elution) solution and
an F-Spin column. Eluted DNA was analyzed using the iTaq universal
SYBR-Green reaction mix (Bio-Rad Laboratories, Inc.). The
methylated primer sets were designed using the MethPrimer tool
(urogene.org/methprimer/). The MSP
primers for methylated and unmethylated regions of the BDP 323 bp
CGI-2 were as follows: M pair (forward,
5'-TTTTTTTAAACGGTTATAGCGAGAC-3' and reverse,
5'-CTTAATTTACCAAATAACCCGTA-3'); and U pair (forward,
5'-TTTTTTAAATGGTTATAGTGAGATGG-3' and reverse,
5'-AATACAACACCTTAATAATCCCAAA-3'). The percentage methylation was
calculated as: 100/[1+2∆Cq (meth-unmeth)]. ∆Cq (meth-unmeth) was
calculated by subtracting the Cq values of methylated CGI signals
from the Cq values of the unmethylated CGI signal (13,14).
Detection of the GLA deletion
variant
The genetic analysis to identify the GLA
mutation was performed by Duke University Health System/BGL-Genzyme
Fabry Testing (Durham). The patient's genomic DNA from peripheral
blood was amplified by PCR, followed by Sanger DNA sequencing of
the coding region of the GLA gene and flanking intronic
sequences, with a minimum of 20 bp of the GLA gene.
α-GAL test
The α-GAL test was performed by Quest diagnostics
(Amarillo, TX, USA) using Flow Injection Analysis on a Tandem Mass
Spectrometry to verify abnormal serum α-GAL results in male
patients with a clinical presentation suggestive of FD (testdirectory.questdiagnostics.com/test/test-detail/37621/alpha-galactosidase-leukocytes?cc=MASTER).
Statistical analysis
Microsoft Excel 365 (Microsoft Corporation) was used
for sorting the data and for analysis. Data are presented as the
mean ± SD. GraphPad Prism version 7.01 (GraphPad Software, Inc.)
was used for statistical analysis of the various parameters
reported in this study. A Student's t-test or a one-way ANOVA with
a post-hoc Tukey's multiple comparisons test was used to evaluate
differences between the independent groups simultaneously and to
test the statistical differences between every possible pair of all
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Case presentation
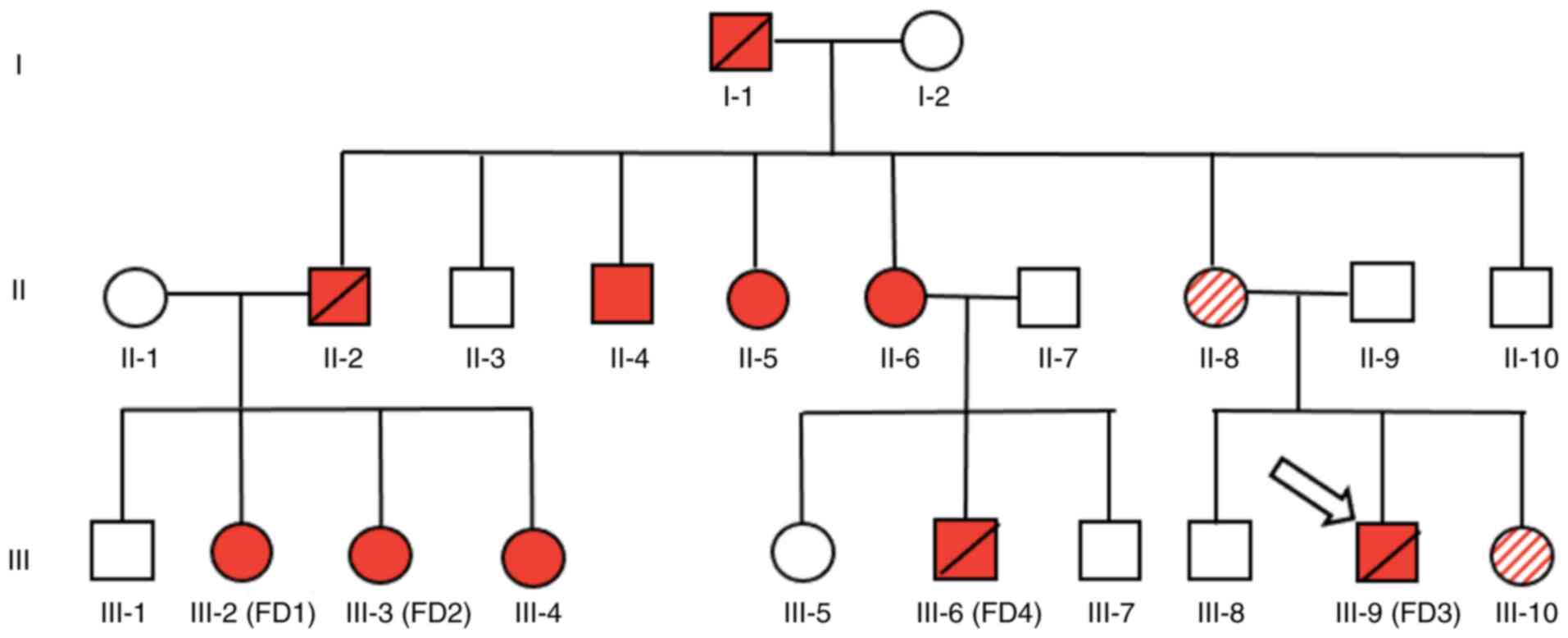
Four members of the same family were diagnosed with
FD (Table I); two males and two
females were recruited for the study from the outpatient clinic of
the Texas Tech University Health Sciences Center. The four patients
showed variable MSSI and FOS-MSSI severity scores (Table SII). The FD family history is shown
in the pedigree diagram (Fig. 1).
Among the male members of the family, one, FD4 (Table I, Fig.
1) was diagnosed based on a very low level of the α-GAL enzyme
(<1%), the test was performed by Quest Diagnostics after
presenting to our clinic with progressive loss of kidney function,
depression, stroke, angiokeratomas, and hypertension. Additionally,
FD3 was diagnosed with the typical distribution of angiokeratomas
and peripheral neuropathy. Two female patients, who were sisters
(FD1 and FD2) had arrhythmias, bradycardia, and peripheral
neuropathy with no renal involvement. BGL-G enzyme Fabry Testing
performed the genetic analysis that showed the presence of a
heterozygote GLA variant c.1033_1034delTC, p.Ser345Argfs
(accession ID: VCV000092538; www.ncbi.nlm.nih.gov/clinvar).
Fabrazyme (agalsidase β), an enzyme replacement
therapy, was used to treat FD patients, which, although prevented
the development of renal involvement in the male who was diagnosed
early, did not lead to the resolution of all symptoms in the
observed patients; the patients were followed-up for 5 years.
Considering our previous finding (12), the potential link between CGI
methylation levels of the BDP and the expression of GLA and
HNRNPH2 was investigated in the FD family members with
variable clinical manifestations and FD severity.
The genomic setting of TC deletion in
the GLA locus
The GLA locus is mapped to the reverse strand
of the RPL36A-HNRNPH2 readthrough locus that appears
on the forward strand of the complete genomic region NC_000023.11
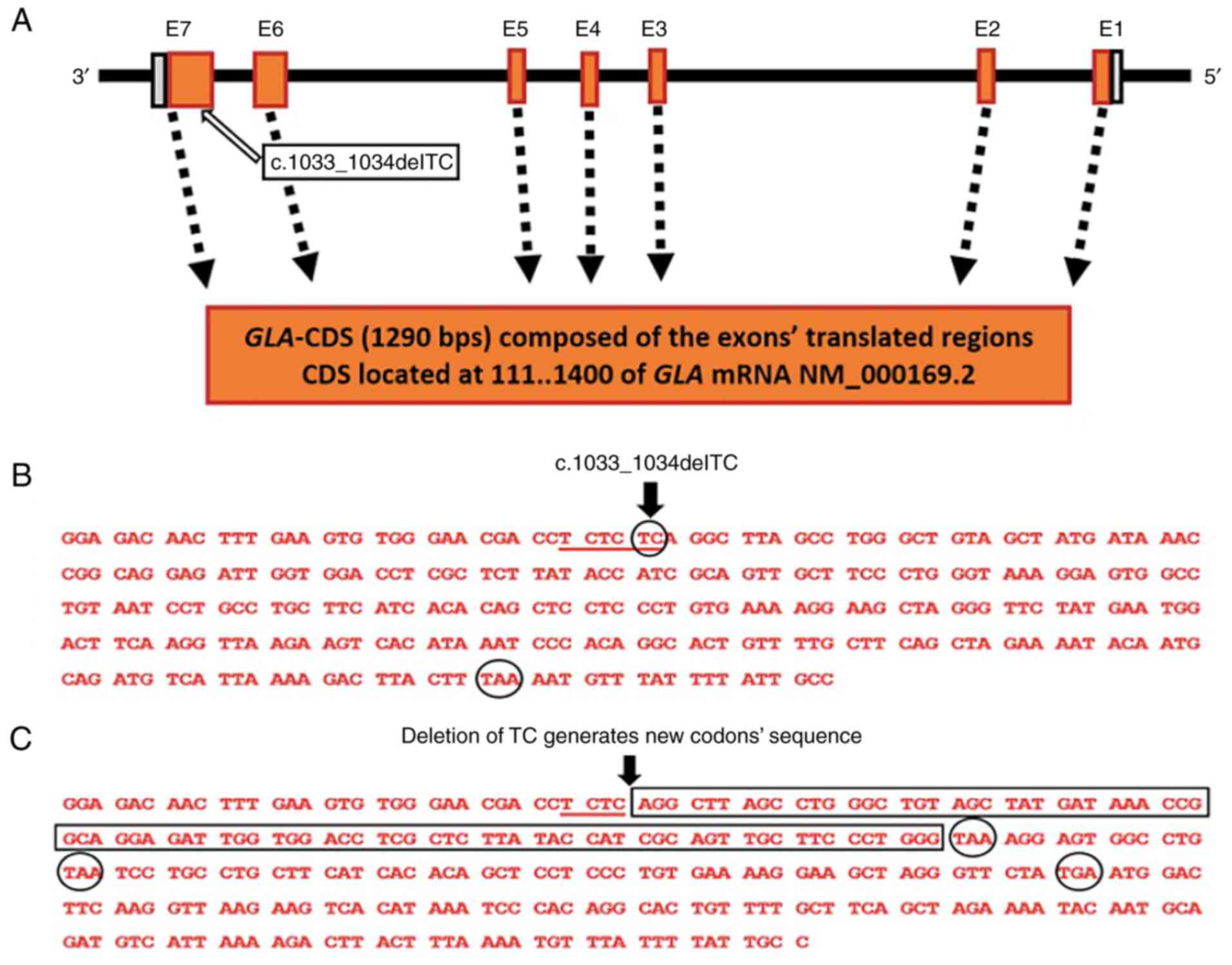
(Fig. 2A). The c.1033_1034delTC
deletion mutation is in GLA exon seven of the translated
region (Fig. 2B and 3A). The position of the TC dinucleotide
deletion is c.1033_1034delTC in the coding sequence at the last TC
dinucleotide of the TC tri-dinucleotide repeat, TCTCTC, underlined
in Fig. 3B. The consequence of TC
deletion is a shift in the DNA sequence and the generation of a
distorted reading frame of the coding sequence and the formation of
three premature nonsense codons indicated by the circled portion of
the sequence in Fig. 3C, compared
with one natural termination codon, TAA in the normal GLA
sequence (Fig. 3B). The deletion
mutation results in an amino acid serine (S) (Fig. 4A), encoded by the triplet TCA
(Fig. 3B), along the α-Gal A
polypeptide sequence, being altered to a new sequence starting with
arginine (R) (Fig. 4B), encoded by
the triplet AGG (Fig. 3C). The new
amino acid sequence is terminated by a premature nonsense codon,
TAA (END) (Fig. 3C and 4B). In addition to the NCBI-ClinVar
database (Table II), peer-reviewed
studies (18-23)
have also reported the deletion variant, c.1033_1034delTC, in FD
patients previously.
| Table IIPrevious submissions on the
c.1033_1034delTC (p.Ser345Argfs) variant.a |
Table II
Previous submissions on the
c.1033_1034delTC (p.Ser345Argfs) variant.a
| Submitter and
submission date | Clinical
significance | Submission
accession |
|---|
| GeneDx, Sep 21,
2015 | Pathogenic | SCV000292562.9 |
| Integrated
Genetics/Laboratory Corporation of America, Jun 3, 2016 | Pathogenic | SCV000695728.1 |
| EGL Genetic
Diagnostics, Eurofins Clinical Diagnostics, Apr 20, 2018 | Pathogenic | SCV000110103.8 |
Methylation analysis of the GLA-
HNRNPH2 BDP
The GLA and HNRNPH2 loci are located
within the readthrough locus RPL36A-HNRNPH2, the GLA
locus is at the reverse strand, whereas the HNRNPH2 locus
appears at the forward strand (Fig.
2A). Our previous study showed one of three identified BDP CpG
islands, CGI-2 composed of 323 bp and mapped along the BDP
sequence, was methylated in four normal human cell types (12). Using the same Methylation-Specific
PCR (MSP) protocol and the same primers reported in our previous
study (12), the methylation status
of the BDP CGI-2 at position 241-563 (Fig. S1) in DNA isolated from blood
samples of FD patients compared with normal individuals was
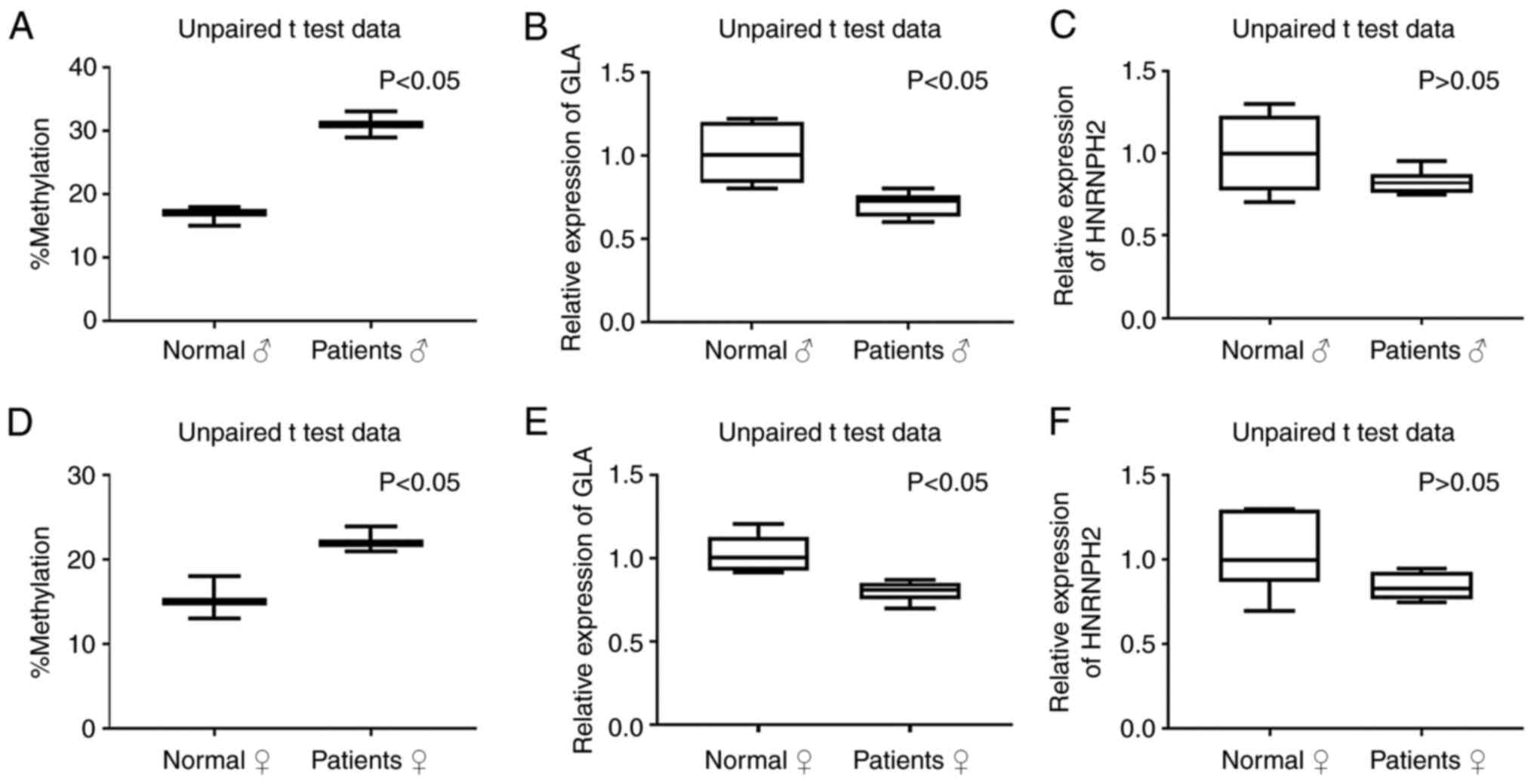
evaluated. The DNA methylation analysis showed variable levels of
methylation in BDP in the tested blood samples; DNA methylation was
elevated in both the male and female patients compared with
methylation in the normal group (Fig.
5A-1 and B-1, P<0.05).
Expression of GLA and HNRNPH2 in Fabry
patients and healthy individuals
The molecular events regulating the expression of
the GLA and HNRNPH2 transcripts are not well
established. As a follow-up to our previous finding of
GLA-HNRNPH2 BDP and the observed methylation levels in
normal kidney and skin cells (12),
in this study, the potential impact of BDP methylation on
GLA and HNRNPH2 expression in four FD patients
carrying the deletion mutation c.1033_1034delTC (p.Ser345Argfs) was
determined. As shown in Fig. 5, the
expression of GLA is significantly lower (P<0.05) and
HNRNPH2 showed a tendency of low expression (P=0.1)) in the
FD patients when the levels of BDP methylation were high compared
with high GLA expression when BDP methylation was low in
normal individuals (P<0.05). The results showed potential
accumulative effects of the GLA mutation c.1033_1034delTC
and BDP methylation with the severity of disease in FD patients as
discussed below. This association was clearly demonstrated in male
patient FD4, a family member who was diagnosed with progressive
loss of kidney function, depression, stroke, angiokeratomas, and
hypertension. He had the highest BDP DNA methylation and lowest
GLA and HNRNPH2 expression levels (Fig. S2, Table SIII).
Discussion
The detailed genetic presentation has not yet been
fully elucidated in FD, which is a clinically heterogeneous, slow,
and progressive disease that can show >70 symptoms (rarediseases.info.nih.gov). Although FD is a
life-threatening, multisystemic condition, and patients exhibit a
wide range of clinical symptoms, the primary cause of the disease
is attributed to pathogenic GLA mutations (1-5).
The present study highlights the association of additional genetic
factors in addition to GLA with FD and shows an indicator of
suspicion that FD is caused solely by GLA mutations. Our
previous study (12) and the
current study show the potential of including study of
HNRNPH2 and the GLA-HNRNPH2 BDP methylation
status in the diagnosis, therapy, and development of the
disease.
Although few studies have dealt with the role of
methylation in FD, for a review see Di Risi et al (24); the present study provided further
evidence on the potential role of DNA methylation involvement in
the clinical manifestation of FD. The GLA mutations can
cause total or partial decreased activity of α-Gal A and
accumulation of glycosphingolipids (3,4).
Intriguingly, the potential source of GLA-HNRNPH2 BDP
methylation is likely sphingolipids. Aside from their prominent
roles as structural lipids, sphingolipids and their metabolizing
enzymes are found in the nucleus and linked to chromatin remodeling
and epigenetic regulation of gene expression (25). Furthermore, additional species of
sphingolipids serve different functions, such as functioning as
signaling molecules and can control gene expression via DNA
methylation (26). A further point
is that the FD patients in the present study were carriers of a TC
deletion caused by c.1033_1034delTC in the GLA exon 7 near
the 3'-UTR region, and such a deletion may influence the regulatory
function of the 3'-UTR sequence. For example, methylation of the
N(6) position of adenosine
m(6)A is a posttranscriptional
modification of RNA. It was found that m(6)A sites are enriched near stop codons and
in 3' UTRs, and there is an association between m(6)A residues and microRNA-binding sites
within 3' UTRs (27).
The majority of patients with FD may experience
chronic or episodic pain, known as FD crises or acroparaesthesiae
(12,28-30).
The development of pain in FD is hypothesized to be primarily
neuropathic; the suggested cause is serum and tissue accumulation
of Gb3 and its influence on the peripheral nervous system, which
may lead to cell swelling (31-34).
Furthermore, a question has been raised on whether the
HNRNPH2 and the BDP methylation may play a role in
diagnosing and treating chronic pain in FD patients and other
related FD clinical symptoms. Earlier studies have demonstrated the
association between alternative RNA splicing and pain (35,36).
The products of HNRNP genes, including HNRNPH2, are RNA
binding proteins that are associated with the mRNA splicing process
(12). HNRNPH1 and
HNRNPF are post-transcriptional regulators of opioid
receptor expression (37), and
similar protein structures are produced by HNRNPH2 and
HNRNPF (38). This may
suggest HNRNPH2 involvement in the pain experienced by FD
patients. BDP methylation may cause abnormalities in HNRNPH2
expression and defects in mRNA splicing. A previous study suggested
that DNA methylation not only affects gene expression but also
regulates alternative splicing (39).
At present, little is known regarding the role of
RPL36A in FD. The knockdown of RPL36A, the first gene
in the RPL36A-HNRNPH2 readthrough region, using a targeting
siRNA showed a significant decrease not only in RPL36A
expression but also in GLA expression (40). The RPL36A gene, also known as
MIG6, encodes the ribosomal protein L36a, and over-expression of
this protein is associated with cellular proliferation in
hepatocellular carcinoma (41,42).
In the present study, we also sought to explain the
heterozygous status of GLA variant c.1033_1034delTC,
p.Ser345Argfs in the FD female patients. The inheritance of several
X-linked conditions is not visibly dominant or recessive (43). In females, one altered copy of the
gene usually leads to less severe health problems than those in
affected males, or it may have no warning signs. In females a high
FD penetrance was observed; at least 70% of females showed the
clinical manifestations of the disease (44). Thus, it is suggested that when
referring to females with FD, the term carrier should be avoided
and replaced by the term heterozygotes (45).
Finally, although the results of the present study
showed further evidence of the potential involvement of BDP
methylation, in addition to the GLA gene and the
HNRNPH2 gene in FD severity, the precise mechanism that
regulates the bidirectional transcription of GLA and
HNRNPH2 is yet to be fully understood. Additional studies
using novel experimental and bioinformatics-based methods,
including high-throughput approaches and data analysis by
developing machine learning models for computational estimation of
methylation profiling (46,47) are required for a better
understanding of the architecture, cis-regulatory elements of
GLA and HNRNPH2 and the cumulative effects of
GLA mutations and the GLA-HNRNPH2 BDP
methylation in FD.
Supplementary Material
Location of the CpG island in the
GLA and HNRNPH2 BDP sequence. The CpG island is
underlined.
BDP methylation and GLA and
HNRNPH2 expression in the normal individuals and FD
patients. (A) DNA methylation of the BDP in normal females (C1 and
C2) and FD females (FD1 and FD2). (B and C) Expression of
GLA and HNRNPH2 in normal females (C1 and C2) and FD
females (FD1 and FD 2). (D) DNA methylation of the BDP in normal
males (C3 and C4) and FD males (FD3 and FD4). (E and F) Expression
of GLA and HNRNPH2 in the normal males (C3 and C4)
and FD males (FD3 and FD4). FD, Fabry disease patients; BDP,
bidirectional promoter.
Genetic conditions associated with
variants in human RPL36A-HNRNPH2 (ENSG00000257529)
readthrough, including the GLA and rat orthologue gene
ENSRNOG00000050463.a
Severity scores of FD patients.
Summary of GLA and
HNRNPH2 expression vs. BPD methylation and severity of FD in
the four patients. Severity score is the sum of the FOS-MSSI scores
per patient in Table I. Methylation
and expression vs. control.
Acknowledgements
Not applicable.
Funding
Funding: This study was funded by Sanofi-Genzyme Corporation
(grant no. GZ-2017-11708).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TLV analyzed and interpreted the clinical patient
data. MAAO and IIAO performed and interpreted the molecular
analyses. TLV and MAAO confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Texas Tech University Health Science Center
(Amarillo, USA).
Patient consent for publication
Written informed consent was obtained from the
healthy individuals and patients, whom all agreed to the use of
their blood samples for scientific research and for the publication
of the anonymized data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mehta A and Hughes DA: Fabry Disease. Adam
MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW and Amemiya
A (eds). GeneReviews® Seattle: University of Washington,
Seattle, WA, 1993-2018, 2017.
|
|
2
|
Garman SC and Garboczi DN: The molecular
defect leading to Fabry disease: Structure of human
alpha-galactosidase. J Mol Biol. 337:319–335. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zarate YA and Hopkin RJ: Fabry's disease.
Lancet. 372:1427–1435. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Svarstad E and Marti HP: The changing
landscape of Fabry disease. Clin J Am Soc Nephrol. 15:569–576.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rozenfeld P and Neumann PM: Treatment of
fabry disease: Current and emerging strategies. Curr Pharm
Biotechnol. 12:916–922. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lidove O, West ML, Pintos-Morell G, Reisin
R, Nicholls K, Figuera LE, Parini R, Carvalho LR, Kampmann C,
Pastores GM and Mehta A: Effects of enzyme replacement therapy in
Fabry disease: A comprehensive review of the medical literature.
Genet Med. 12:668–679. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Keating GM: Agalsidase alfa: A review of
its use in the management of Fabry disease. BioDrugs. 26:335–354.
2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thurberg BL, Rennke H, Colvin RB, Dikman
S, Gordon RE, Collins AB, Desnick RJ and O'Callaghan M:
Globotriaosylceramide accumulation in the Fabry kidney is cleared
from multiple cell types after enzyme replacement therapy. Kidney
Int. 62:1933–1946. 2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Khanna R, Soska R, Lun Y, Feng J,
Frascella M, Young B, Brignol N, Pellegrino L, Sitaraman SA,
Desnick RJ, et al: The pharmacological chaperone
1-deoxygalactonojirimycin reduces tissue globotriaosylceramide
levels in a mouse model of Fabry disease. Mol Ther. 18:23–33.
2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Germain DP, Hughes DA, Nicholls K, Bichet
DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F,
Amartino H, et al: Treatment of Fabry's disease with the
pharmacologic chaperone migalastat. N Engl J Med. 375:545–555.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Modrego A, Amaranto M, Godino A, Mendoza
R, Barra JL and Corchero JL: Human α-galactosidase A mutants:
Priceless tools to develop novel therapies for Fabry disease. Int J
Mol Sci. 22(6518)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Al-Obaide MA, Al-Obaidi II and Vasylyeva
TL: Unexplored regulatory sequences of divergently paired GLA and
HNRNPH2 loci pertinent to Fabry disease in human kidney and skin
cells: Presence of an active bidirectional promoter. Exp Ther Med.
21(154)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ng EK, Leung CP, Shin VY, Wong CL, Ma ES,
Jin HC, Chu KM and Kwong A: Quantitative analysis and diagnostic
significance of methylated SLC19A3 DNA in the plasma of breast and
gastric cancer patients. PLoS One. 6(e22233)2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shaker MM, Shalabi TA and Amr KS:
Correlation of methylation status in MTHFR promoter region with
recurrent pregnancy loss. J Genet Eng Biotechnol.
19(44)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Whybra C, Bähner F and Baron K:
Measurement of disease severity and progression in Fabry disease.
In: Fabry Disease: Perspectives from 5 Years of FOS. Mehta A, Beck
M and Sunder-Plassmann G (eds). Chapter 32. Oxford: Oxford
PharmaGenesis, 2006.
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ashley GA, Shabbeer J, Yasuda M, Eng CM
and Desnick RJ: Fabry disease: Twenty novel alpha-galactosidase A
mutations causing the classical phenotype. J Hum Genet. 46:192–196.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Germain D, Biasotto M, Tosi M, Meo T, Kahn
A and Poenaru L: Fluorescence-assisted mismatch analysis (FAMA) for
exhaustive screening of the alpha-galactosidase A gene and
detection of carriers in Fabry disease. Hum Genet. 98:719–726.
1996.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lukas J, Giese AK, Markoff A, Grittner U,
Kolodny E, Mascher H, Lackner KJ, Meyer W, Wree P, Saviouk V and
Rolfs A: Functional characterisation of alpha-galactosidase a
mutation as a basis for a new classification system in fabry
disease. PLoS Genet. 9(e1003632)2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nakano S, Morizane Y, Makisaka N, Suzuki
T, Togawa T, Tsukimura T, Kawashima I, Sakuraba H and Shibasaki F:
Development of a highly sensitive immuno-PCR assay for the
measurement of α-galactosidase A protein levels in serum and
plasma. PLoS One. 8(e78588)2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rodríguez-Marí A, Coll MJ and Chabás A:
Molecular analysis in Fabry disease in Spain: Fifteen novel GLA
mutations and identification of a homozygous female. Hum Mutat.
22(258)2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shabbeer J, Yasuda M, Benson SD and
Desnick RJ: Fabry disease: Identification of 50 novel
alpha-galactosidase A mutations causing the classic phenotype and
three-dimensional structural analysis of 29 missense mutations. Hum
Genomics. 2:297–309. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Di Risi T, Vinciguerra R, Cuomo M, Della
Monica R, Riccio E, Cocozza S, Imbriaco M, Duro G, Pisani A and
Chiariotti L: DNA methylation impact on Fabry disease. Clin
Epigenetics. 13(24)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Spiegel S, Milstien S and Grant S:
Endogenous modulators and pharmacological inhibitors of histone
deacetylases in cancer therapy. Oncogene. 31:537–551.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Silva GD, Coeli-Lacchini FB and Leopoldino
AM: How do sphingolipids play a role in epigenetic mechanisms and
gene expression? Epigenomics. 14:219–222. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3' UTRs and near stop codons. Cell.
149:1635–1646. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Magg B, Riegler C, Wiedmann S, Heuschmann
P, Sommer C and Üçeyler N: Self-administered version of the
Fabry-associated pain questionnaire for adult patients. Orphanet J
Rare Dis. 10(113)2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gibas AL, Klatt R, Johnson J, Clarke JT
and Katz J: A survey of the pain experienced by males and females
with Fabry disease. Pain Res Manag. 11:185–192. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Crosbie TW, Packman W and Packman S:
Psychological aspects of patients with Fabry disease. J Inherit
Metab Dis. 32:745–753. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tabira T, Goto I, Kuroiwa Y and Kikuchi M:
Neuropathological and biochemical studies in Fabry's disease. Acta
Neuropathol. 30:345–354. 1974.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gadoth N and Sandbank U: Involvement of
dorsal root ganglia in Fabry's disease. J Med Genet. 20:309–312.
1983.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Miller JJ, Aoki K, Moehring F, Murphy CA,
O'Hara CL, Tiemeyer M, Stucky CL and Dahms NM: Neuropathic pain in
a Fabry disease rat model. JCI Insight. 3(e99171)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rajan JN, Ireland K, Johnson R and Stepien
KM: Review of mechanisms, pharmacological management, psychosocial
implications, and holistic treatment of pain in Fabry disease. J
Clin Med. 10(4168)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Donaldson LF and Beazley-Long N:
Alternative RNA splicing: Contribution to pain and potential
therapeutic strategy. Drug Discov Today. 21:1787–1798.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
de la Peña JB and Campbell ZT:
RNA-binding proteins as targets for pain therapeutics. Neurobiol
Pain. 4:2–7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Song KY, Choi HS, Law PY, Wei LN and Loh
HH: Post-transcriptional regulation of mu-opioid receptor: Role of
the RNA-binding proteins heterogeneous nuclear ribonucleoprotein H1
and F. Cell Mol Life Sci. 69:599–610. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Alkan SA, Martincic K and Milcarek C: The
hnRNPs F and H2 bind to similar sequences to influence gene
expression. Biochem J. 393:361–371. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lev Maor G, Yearim A and Ast G: The
alternative role of DNA methylation in splicing regulation. Trends
Genet. 31:274–280. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Al-Obaide MA and Vasylyeva TL: The
Knockdown of RPL36A downregulates GLA expression associated with
Fabry disease in vitro model. ASN ePosters. (Abstract
PO1600)2020.
|
|
41
|
Kim JH, You KR, Kim IH, Cho BH, Kim CY and
Kim DG: Over-expression of the ribosomal protein L36a gene is
associated with cellular proliferation in hepatocellular carcinoma.
Hepatology. 39:129–138. 2004.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Xu M, Wang Y, He HT and Yang Q: MiR-589-5p
is a potential prognostic marker of hepatocellular carcinoma and
regulates tumor cell growth by targeting MIG-6. Neoplasma.
65:753–761. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Happle R: X-chromosome inactivation: Role
in skin disease expression. Acta Paediatr Suppl. 95:16–23.
2006.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Dobyns WB: The pattern of inheritance of
X-linked traits is not dominant or recessive, just X-linked. Acta
Paediatr Suppl. 95:11–15. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Rozenfeld PA: Fabry disease: Treatment and
diagnosis. IUBMB Life. 61:1043–1050. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Weingarten-Gabbay S, Nir R, Lubliner S,
Sharon E, Kalma Y, Weinberger A and Segal E: Systematic
interrogation of human promoters. Genome Res. 29:171–183.
2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Fan S, Wang L, Liang L, Cao X, Tang J and
Tian Q: The progress on the estimation of DNA methylation level and
the detection of abnormal methylation. Quant Biol. 10:55–66.
2022.
|