1. Introduction
Neurodegenerative disorders have a exhibited a
marked increase in incidence worldwide, thus rendering them a
primary concern for the scientific society. The genetic cause of
numerous disorders has already been described (1-3)
and, nowadays, research focuses on the timely diagnosis and
effective therapy of the most common neurodegenerative disorders,
such as Parkinson's disease, Alzheimer's disease, amyotrophic
lateral sclerosis and Huntington's disease (HD). These disorders
have diverse clinical manifestations; however, some of them
demonstrate similarities among patients (4,5).
Although in numerous cases, the onset of neurodegenerative
disorders appears in middle to late adult life, there are patients
who manifest symptoms of these disorders at a very early age
(6,7). HD is one of the most common disorders
with severe symptomatology, which affects individuals of all ages,
progressively leading to severe disabilities. The genetic basis of
this disorder has been established and has been known for a few
decades now, and recent research has revealed promising mechanisms
for eliminating HD symptoms (8).
Furthermore, HD can be regarded as a model neurodegenerative
disorder for the study of other cases with shared symptoms, and
knowledge of other diseases may be useful for HD diagnosis and
treatment.
2. Genetics and pathology of Huntington's
disease
HD is a fatal, autosomal dominant, progressive
neurodegenerative disorder characterized by severe symptoms,
including motor, cognitive and psychiatric symptoms, atrophy of the
basal ganglia and the cerebral cortex, and an inevitably
progressive course, resulting in mortality 5-20 years following the
manifestation of symptoms. Typically, the motor defects include
chorea and loss of coordination, and patients also demonstrate
difficulty with speech and swallowing (9). Cognitive symptoms can be detected up
to a decade prior to diagnosis and cognitive ability declines as
the disease progresses (10).
Psychiatric symptoms, such as depression, psychosis and
obsessive-compulsive disorder, are also common in HD and are
particularly distressing for patients (11,12).
Patients with HD eventually require a wheelchair and more severe
symptoms may lead to them becoming bedridden, with all the
complications that may derive from that form of immobility.
From a neuropathological point of view, in patients
with HD, the dysfunction and death of specific neurons within their
brains are observed. There is a wide range in the age of onset of
HD, as both juveniles (13,14) and adults have been diagnosed with
the disorder thus far. For instance, kindred members of families
that revealed a history consistent with HD autosomal dominant
inheritance, took part in a 20-year study, which was published in
2004(15). The researchers of that
study found that the typical ages of disease onset were between 21
and 50 years of age (15). Although
the disorder typically manifests in adulthood, juvenile HD (JHD) is
also frequent among patients (16).
A recent study, conducted in Argentina in 2015(17), reported that almost 20% of the
patients diagnosed with HD, revealed their first symptoms of the
disorder during their childhood. It should be noted that the
overall estimated prevalence of JHD of that study was higher than
that in any other population recorded to date (17). The brain structure in young patients
was previously assessed by Tereshchenko et al (18) in 2019, proposing that the morphology
differs among juveniles and adult patients, as young patients
revealed proportional cerebellar enlargement (18). In the same year, another study
suggested that the pathogenesis of HD begins with abnormal brain
development in both child and adolescent patients (19).
The first attempt to discover the genetic cause for
HD by Gusella et al (20)
revealed that the HD gene is linked to a polymorphic DNA marker
that maps to human chromosome 4, in particular 4p16.3. A decade
later, a Huntington's Disease Collaborative Research Group
discovered that mutations in the Huntington (HTT) gene encoding the
huntingtin protein, a large protein of 3,144 amino acids, led to
the neurodegenerative disorder (21). In particular, they suggested that
the disorder is caused due to a cytosine-adenine-guanine (CAG)
trinucleotide expansion in exon 1 that codes for polyglutamine
(polyQ) in the N-terminal of the HTT gene (21). The CAG sequence is normally repeated
9 to 35 times, with an average median of between 17 and 20 repeats.
However, patients with HD usually reveal a CAG expansion exceeding
35 repeats (22). Above a threshold
of ~35 CAG repeats, the age of onset of HD is inversely associated
with the length of the expansion. A recent study conducted by
Schultz et al (23)
demonstrated that the development of verbal skills appeared to
plateau earlier as CAG repeat length increased. The repeats are
usually between 36 and 39, depending on the age. Juveniles with HD
exhibit high repeat lengths (24).
In certain rare cases, patients exhibit less repeats in their
genome, 27-35, demonstrating an endophenotype (25).
Of note, the huntingtin protein is expressed in all
cell types of the body, both at the tissue and subcellular level,
in all developmental stages. Recently, research has focused on the
investigation of the HTT structure via cryo-electron microscopy
contributing to a better comprehension of its morphology and form
abnormalities (26). It has been
described as a 350-kDa HEAT-repeat protein which interacts with
hundreds of other proteins (27)
and participates in numerous cellular processes. Although the
cellular functions of HTT protein are not yet completely
understood, it appears to play a crucial role during early
embryonic development and neurogenesis. In particular, Saudou and
Humbert (22) described the human
huntingtin protein sequence and its post-translational
modifications in detail. They suggested that it coordinates cell
division, as it participates in the proper mitotic spindle
positioning and it regulates ciliogenesis. They also noted that
huntingtin mediates endocytosis, vesicle recycling and endosomal
trafficking, as it interacts with other proteins which are related
to these mechanisms. Other functions of the protein include
autophagy and transcription (22).
It should be underlined that the HTT gene physiological
expression is essential for organism homeostasis as it plays a
neuroprotective role as well, even against mutant HTT (mHtt)
toxicity (28,29). Moreover, HTT protein plays a crucial
role in mitochondrial structure and function in the embryogenesis
and oxidative metabolism, and HTT mutations have been linked to
mitochondrial abnormalities (30-32).
The role of both wild-type Htt and mHtt in gene
silencing studies has been investigated for the development of an
effective therapy. As regards mHtt molecules, they form toxic
aggregates into the central nervous system, depending on the length
of polyQ expansion. For instance, mHtt co-aggregates with other
proteins which play a crucial role in the cell, leading to
misfunctioned phenotypes. Numerous studies have focused on the
effects of HTT gene knockouts and knockdowns in cellular
function. For example, a study published in 2017 suggested that
mutations in HTT protein are related to nucleocytoplasmic transport
disruption, leading to the improper function of cells (33). During initial experiments performed
on mice with HTT knockdown mutants, the mice succumbed after
8 days of gestation (34). Other
studies have demonstrated that HTT deletion in the mouse
central nervous system leads to a phenotype similar to that of HD
(35,36). It is worth mentioning that a recent
study suggested that HTT variants are also linked to another
disorder with similar symptomatology with HD, the so-called
Lopes-Maciel-Rodan syndrome (37).
In addition, other studies have revealed the ability of various
molecular chaperones, such as the heat shock family proteins,
HSP40, HSP70, HSP90 and HSP105, to combine with misfolded mHtt and
inhibit aggregate formation, leading to cell survival (38,39).
It is clear that the loss of HTT function
contributes to HD pathology and for this reason, it is essential
for survival. The reduction of mHtt levels should be accompanied by
regular HTT expression.
3. Diagnosis and genetic counseling
It has already been mentioned that the age of onset
of HD is inversely associated with the length of the expansion in
the HTT gene. For instance, rare carriers of 36 to 39 CAG
repeats have lower penetrance and a later onset of the disease than
those with 40 or more CAG repeats. Additionally, Keum et al
(40) found that, along with
clinical onset, the age of patients with HD at the time of death
was well determined by an expanded CAG-repeat length. However, they
claimed that the overall duration of the disease was independent of
the length of the mutation's (40).
These data may be useful, not only for the molecular diagnosis of
the disorder, but also for the prediction of the outset of HD
symptomatology. For the molecular diagnosis of the disorder,
various PCR methods have been demonstrated in order to detect CAG
expansions (41). A recent study
presented a novel triplet-primed PCR-based assay aiming to improve
the test reliability and accuracy by detecting CAG expansions in
samples with sequence variations in the HTT gene (42).
It is known that miRNAs are involved in the
biological processes of development, proliferation, inflammation
and apoptosis, and their expression has been linked to HD diagnosis
and symptomatology. For instance, Langfelder et al (43) found that the abnormal expression of
miRNAs played a critical role in HD pathogenesis. For this reason,
apart from the direct quantification of mHTT itself, which is the
main disease-related biomarker, other miRNAs may be useful tools as
biomarkers for HD prognosis (44).
Furthermore, numerous diagnostic tests have been
proposed thus far, based on criteria related to inheritance and the
symptomatology of the individual; however, these methods need to be
improved. Patients who experience certain cognitive and behavioral
symptoms may have HD (45,46). A recent study proposed the Enroll-HD
dataset for estimating disease onset and its diagnostic confidence
level (47). The results of that
study were not promising, suggesting that it is important to
develop more reliable diagnostic criteria (47). Another diagnostic approach suggested
that the concentration of trace elements in the blood of patients
with HD differs from that of healthy individuals. Researchers found
increased levels of the essential elements iron, chromium, selenium
and zinc and of the non-essential element, arsenic, in the blood of
patients with HD, suggesting that the blood metal profile may be
used as an easy tool for the disorder's medical detection (48).
HD follows the Gregor Mendel's principles of
inheritance, as it is inherited in an autosomal-dominant manner.
The offspring of an individual with a pathogenic variant,
heterozygote, have a 50% chance of inheriting the disease-causing
allele. Genetic counseling includes predictive testing in
asymptomatic adults and prenatal testing in order to reveal the
mutated allele (49). The
prevalence of HD is ~1 in 10,000 individuals in the USA, as well as
in Europe (50,51). In the year 2000, Sobel and Cowan
(52) conducted predictive testing
on asymptomatic individuals at risk of developing HD in the context
of genetic counseling. Family members were requested to describe
their communication and interactions with the social environment,
and provide concerns about their future care. Members in 50% of the
families experienced changes in patterns of communication and 56%
of the participants reported changes in current relationships. The
researchers suggested that families may benefit in pretest sessions
by examining their patterns of dealing with illness issues, both
past and present (52).
Migliore et al (53) suggested different approaches of
counseling, depending on the genetic condition of the individual.
For instance, in the case of intermediate alleles (27-35 CAG
repeats) the experts should explain the potential risk of mutations
and other members of the family should also be tested. In the case
of low penetrance alleles (36-39 CAG repeats), individuals should
be informed about the risk of HD symptoms manifesting at any age.
Counseling for all family members is also required when juveniles
are diagnosed with JHD. When the HD mutation is detected in a
prenatal genetic test, the parents should be informed for the risk
of the newborn manifesting the disease and should be given the
option of terminating the pregnancy (53). This approach utilizes the current
knowledge of the molecular basis of HD with the inclusive genetic
counseling of all relatives. Recently, MacLeod et al
(54) proposed a family systems
approach to genetic counseling, which uses the narrative model.
With the narrative resources, the genetic counselor can contribute
to generate new meanings that the person may give to their
experience of the genetic condition and help the patient adapt to
living with the disorder or its risks (54). Another interesting comparative
study, that was performed over the past two decades, on how parents
inform their children who are at risk about their genetic risk
demonstrated that, although testing is performed more often, the
overall attitude towards information and testing has not changed
significantly (55). This
ascertainment proposes that new methods for more comprehensible
information and accessible genetic counseling need be
developed.
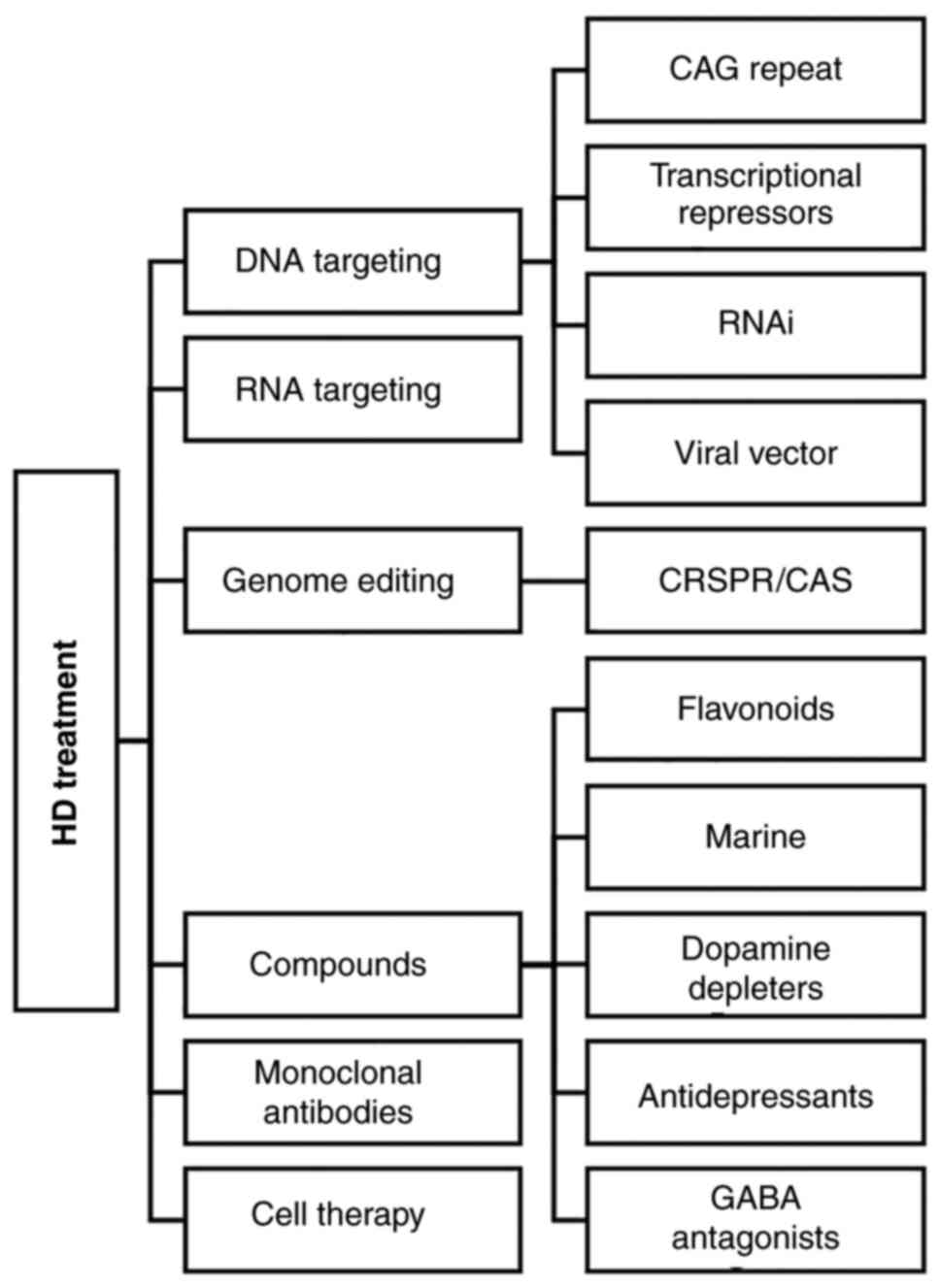
4. Treatment
Research focusing on understanding the underlying
molecular mechanisms leading to the HTT gene mutations is
highly promising, aiming to find a cure for HD. However, current
treatments for HD are still limited. The therapies applied focus on
the treatment of symptoms, as neuroprotective therapies to prevent
disease onset and to attenuate the progression of the disease are
not yet available. For instance, it has been proven that HD is
caused by toxic properties of mHTT, rather than merely the decrease
of wild-type HTT; for this reason, approaches focusing on mHTT
expression, such as lowering HTT mRNA and mutant huntingtin
protein, appear to be promising (56). The main strategies which have been
demonstrated thus far as treatments for HD are presented in
Fig. 1.
A recent study proposed that targeting of CAG
repeat-dependent mechanisms, through gene-silencing approaches, may
affect the rate of functional, motor and cognitive impairment, but
not weight loss, in manifest HD mutation carriers (57). The standard approaches to DNA
targeting use some form of specific DNA-binding element combined
with nucleases, epigenetic modulators, or transcription factors.
Zinc-finger transcriptional repressor approaches may lower mHTT
levels by targeting DNA without altering it, whereas zinc-finger
nucleases can add to the repressive effect of Zinc-finger proteins
that reduce the levels of gene expression by simply binding to DNA
and preventing gene transcription by actually disrupting or
correcting the mutant gene (58).
Along with similar techniques to other direct genome editing
strategies, such as CRISPR/Cas9, strategies that are targeted in
lowering huntingtin and HTT genome editing have immense potential
for the treatment of HD. The main advantage is the permanent
correction of the disease-causing CAG expansion. An antisense
mechanism targeting HTT RNA, using synthetic antisense
oligonucleotides (ASOs) that bind to the specific sequence of
ribonucleic acid, may reduce the mRNA translation to the HTT
disease-causing protein (59). The
ASOs are widely distributed throughout the central nervous system
and they do not require a viral or lipid carrier, resulting to an
effective and simple to develop treatment. Tabrizi et al
(60) used the antisense
oligonucleotide IONIS-HTTRx designed to inhibit HTT mRNA by
triggering the RNase H1-mediated degradation of the target mRNA, in
order to minimize the concentration of mutant huntingtin in
cerebrospinal fluid. They conducted an extended research with 34
patients who were treated with increasing dose levels of 10 to 120
mg and the observations were compared with individuals who received
the placebo. The results revealed that the reduction in
concentrations of mutant huntingtin was dose-dependent (60). The strategy for post-transcriptional
gene suppression using non-coding double-stranded RNA sequences is
known as the RNA interference (RNAi) mechanism. The RNAi pathway
has been used thus far to suppress specific genes of interest and
the results are highly promising for numerous diseases. There are
various molecules which can be used for this purpose, such as
siRNAs, shRNAs and artificial miRNAs that have been used to
eliminate the HD symptoms. The first trials were performed two
decades ago in rodents. In 2005, Harper et al (61) used a shRNA molecule to target the
HTT mutant gene and the results were satisfactory, as the
reduction in mHTT synthesis, prevented inclusions, gait deficits
and rotarod dysfunction. In another case, a siRNA molecule was
injected into the mouse striatum, and the reduction in mHTT
synthesis prolonged striatal neuron survival, reduced aggregates
and prevented motor dysfunction (62). The application of siRNA approaches
has been successful in multiple animal systems (63). In a recent study for example, a
single-stranded siRNA (ss-siRNA) was used for RNAi, resulting in a
selective decrease of CAG-expanded HTT protein in various regions
throughout the mouse brain (64).
Other ribonucleic acids, such as miRNAs have been used for the
suppression of mHTT in genetically modified mice and the results
have been promising; in one case, this strategy led to the
prevention of regional cortical and striatal atrophy, and reduced
weight loss (65). It is worth
mentioning that the majority of the previous technics, apart from
gene editing, effectively reduce, but do not completely eliminate
the production of mHTT.
In addition, the therapeutic approach of
overexpressing wild-type HTT has been investigated. Early trials of
inserting the wild-type HTT into mammalian cells which expressed
mHTT have led to reduced cell death (26).
Numerous research studies have used viral vectors,
such as adeno-associated virus, which encapsulate the RNA
molecules, and their genome is combined with enhancers and
promoters, in order to deliver these agents by injection into the
body (66).
Different compounds may be candidates for the
treatment of neurodegenerative disorders, including HD. A recent
study proposed the utilization of flavonoids, which may reduce
cellular stress and play an anti-inflammatory and anti-apoptotic
role in the cell (67). In another
case, researchers proposed that marine compounds may be used for
the treatment of various neurodegenerative diseases, as they also
demonstrate antioxidant, anti-inflammatory and anti-apoptotic
properties. Tetrabenazine (TBZ), which is an inhibitor that blocks
dopamine uptake into vesicles, has been shown to exert antichorea
effects in patients with HD and was the first approved drug for
medication (68). Since then,
studies on the optimization of drug delivery and bioavailability of
TBZ in patients have been conducted based on latest nanotechnology
technics (69). Several molecules
have been suggested for the treatment of Parkinson's disease, such
as fucoidan and xyloketal B, and fucoxanthin and cerebrosides for
Alzheimer's disease, and have also been investigated for other
disorders, such as HD for effective treatment (70). In 2020, Jabłońska et al
(71) suggested that pridopidine, a
dopamine stabilizer, may be a promising drug for HD symptoms. It is
well-known that there is an association between the amount of
dopamine in the central nervous system and the stage of the
disease, as the causes of HD are dopaminergic conduction disorders,
and experiments on animal models have demonstrated the protective
effect of pridopidine on nerve cells (72,73).
Τhe main advantage of drug treatment for HD is that the
effectiveness and tolerance of each active compound is well-studied
for other neurodegenerative diseases with similar symptomatology.
Thus, it is easier to design a suitable medication personalized to
patient diagnostics. Another study proposed that the application of
monoclonal antibody, which targets the HTT protein may deplete its
concentration in the cell, proving that monoclonal antibodies can
interfere with the pathological processes of mHTT spreading in
vivo (74).
Cell replacement therapy for HD using stem cells may
be another opportunity to alleviate symptoms in patients (75). Furthermore, some case studies have
indicated that exercise and physical activity may be beneficial for
patients in terms of motor function, gait speed and balance, and
social benefits have been also identified (76). Thus, exercise may play a
complementary role in the treatment of the disorder.
5. Conclusions and future perspectives
HD is the first trinucleotide disease that was
described and the first autosomal-dominant disease with a possible
diagnosis prior to the manifestation of symptoms. Since 1983 and
the localization of the gene, knowledge of the disorder has
markedly increased, which is necessary in order to improve the
quality of life of patients and improve therapeutic strategies by
discovering novel molecular targets. The pathophysiology of HD is
significant for designing and developing proper treatments
(77). Science offers possibilities
for attenuating the symptoms of the disease, and even the onset;
however, it is also critical to identify effective biomarkers that
may help prevent HD manifestation by early detection and blocking
its course. Modern therapeutic trial design also vastly relies on
identifying and examining biomarkers relevant to each disease.
The next step may be to evaluate and use data from
genome-wide association studies and account for their clinical
utility. Studies (as aforementioned) towards this direction have
contributed to the existing knowledge concerning the association of
genetic variations to the onset of symptoms and the progression of
HD. The combination of early testing in order to predict the
possible HD onset and new targeted and personalized medicine
represents the future in preventing and hopefully, eliminating
neurodegenerative diseases. The path for science ahead to help
patients with HD is a long one. Until then, finding the optimal
care for patients and caregivers is significant.
Acknowledgements
Not applicable.
Funding
Funding: The authors would like to acknowledge funding from the
following organizations: i) AdjustEBOVGP-Dx (RIA2018EF-2081):
Biochemical Adjustments of native EBOV Glycoprotein in Patient
Sample to Unmask target Epitopes for Rapid Diagnostic Testing. A
European and Developing Countries Clinical Trials Partnership
(EDCTP2) under the Horizon 2020 ‘Research and Innovation Actions’
DESCA; ii) ‘MilkSafe: A novel pipeline to enrich formula milk using
omics technologies’, a research co-financed by the European
Regional Development Fund of the European Union and Greek national
funds through the Operational Program Competitiveness,
Entrepreneurship and Innovation, under the call
RESEARCH-CREATE-INNOVATE (project code: T2EDK-02222); iii)
‘INSPIRED-The National Research Infrastructures on Integrated
Structural Biology, Drug Screening Efforts and Drug Target
Functional Characterization’ (Grant MIS 5002550) implemented under
the Action ‘Reinforcement of the Research and Innovation
Infrastructure’, funded by the Operational Program
‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014-2020)
and co-financed by Greece and the European Union (European Regional
Development Fund), and iv) ‘OPENSCREENGR An Open-Access Research
Infrastructure of Chemical Biology and Target-Based Screening
Technologies for Human and Animal Health, Agriculture and the
Environment’ (Grant MIS 5002691), implemented under the Action
‘Reinforcement of the Research and Innovation Infrastructure’,
funded by the Operational Program ‘Competitiveness,
Entrepreneurship and Innovation’ (NSRF 2014-2020) and co-financed
by Greece and the European Union (European Regional Development
Fund).
Availability of data and materials
Not applicable.
Authors' contributions
All authors (AMP, EP, RG, MS, TM, LP, ID, KP, KD,
DAS, FB, GPC, EE and DV) contributed to the conceptualization,
design, writing, drafting, revising, editing and reviewing of the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Recabarren D and Alarcon M: Gene networks
in neurodegenerative disorders. Life Sci. 183:83–97.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pihlstrøm L, Wiethoff S and Houlden H:
Genetics of neurodegenerative diseases: An overview. Handb Clin
Neurol. 145:309–323. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Shen T, You Y, Joseph C, Mirzaei M,
Klistorner A, Graham SL and Gupta V: BDNF polymorphism: A review of
its diagnostic and clinical relevance in neurodegenerative
disorders. Aging Dis. 9(523)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Guzman-Martinez L, Maccioni RB, Andrade V,
Navarrete LP, Pastor MG and Ramos-Escobar N: Neuroinflammation as a
common feature of neurodegenerative disorders. Front Pharmacol.
10(1008)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ma MW, Wang J, Zhang Q, Wang R, Dhandapani
KM, Vadlamudi RK and Brann DW: NADPH oxidase in brain injury and
neurodegenerative disorders. Mol Neurodegener. 12(7)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mendez MF: Early-onset Alzheimer disease
and its variants. Continuum (Minneap Minn). 25:34–51.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bakels HS, Roos RAC, van Roon-Mom WMC and
de Bot ST: Juvenile-Onset huntington disease pathophysiology and
neurodevelopment: A review. Mov Disord. 37:16–24. 2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kumar A, Kumar V, Singh K, Kumar S, Kim
YS, Lee YM and Kim JJ: Therapeutic advances for Huntington's
disease. Brain Sci. 10(43)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yanagisawa N: The spectrum of motor
disorders in Huntington's disease. Clin Neurol Neurosurg. 94
(Suppl):S182–S184. 1992.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bonner-Jackson A, Long JD, Westervelt H,
Tremont G, Aylward E and Paulsen JS: PREDICT-HD Investigators and
Coordinators of the Huntington Study Group. Cognitive reserve and
brain reserve in prodromal Huntington's disease. J Int Neuropsychol
Soc. 19:739–750. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Harrington DL, Liu D, Smith MM, Mills JA,
Long JD, Aylward EH and Paulsen JS: Neuroanatomical correlates of
cognitive functioning in prodromal Huntington disease. Brain Behav.
4:29–40. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Epping EA, Mills JA, Beglinger LJ,
Fiedorowicz JG, Craufurd D, Smith MM, Groves M, Bijanki KR, Downing
N, Williams JK, et al: Characterization of depression in prodromal
Huntington disease in the neurobiological predictors of HD
(PREDICT-HD) study. J Psychiatr Res. 47:1423–1431. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
van Dijk JG, van der Velde EA, Roos RA and
Bruyn GW: Juvenile huntington disease. Hum Genet. 73:235–239.
1986.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Siesling S, Vegter-van der Vlis M and Roos
RA: Juvenile Huntington disease in the Netherlands. Pediatr Neurol.
17:37–43. 1997.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wexler NS, Lorimer J, Porter J, Gomez F,
Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA,
Gayán J, et al: Venezuelan kindreds reveal that genetic and
environmental factors modulate Huntington's disease age of onset.
Proc Natl Acad Sci USA. 101:3498–3503. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Quarrell OWJ, Brewer HM, Squitieri F,
Barker R, Nance MA and Lanswehrmeyer BG: ‘Juvenile Huntington's
Disease (and other trinucleotide repeat disorders)’. Oxford
University Press, Oxford, 2009.
|
|
17
|
Gatto EM, Parisi V, Etcheverry JL,
Sanguinetti A, Cordi L, Binelli A, Persi G and Squitieri F:
Juvenile Huntington disease in Argentina. Arq Neuropsiquiatr.
74:50–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tereshchenko A, Magnotta V, Epping E,
Mathews K, Espe-Pfeifer P, Martin E, Dawson J, Duan W and Nopoulos
P: Brain structure in juvenile-onset Huntington disease. Neurology.
92:e1939–e1947. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
van der Plas E, Langbehn DR, Conrad AL,
Koscik TR, Tereshchenko A, Epping EA, Magnotta VA and Nopoulos PC:
Abnormal brain development in child and adolescent carriers of
mutant huntingtin. Neurology. 93:e1021–e1030. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gusella JF, Wexler NS, Conneally PM,
Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR,
Sakaguchi AY, et al: A polymorphic DNA marker genetically linked to
Huntington's disease. Nature. 306:234–238. 1983.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
A novel gene containing a trinucleotide
repeat that is expanded and unstable on Huntington's disease
chromosomes. The Huntington's Disease Collaborative Research Group.
Cell. 72:971–983. 1993.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Saudou F and Humbert S: The biology of
Huntingtin. Neuron. 89:910–926. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Schultz JL, van der Plas E, Langbehn DR,
Conrad AL and Nopoulos PC: Age-Related cognitive changes as a
function of CAG repeat in child and adolescent carriers of mutant
Huntingtin. Ann Neurol. 89:1036–1040. 2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Andrew SE, Goldberg YP, Kremer B, Telenius
H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA,
et al: The relationship between trinucleotide (CAG) repeat length
and clinical features of Huntington's disease. Nat Genet.
4:398–403. 1993.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Squitieri F and Jankovic J: Huntington's
disease: How intermediate are intermediate repeat lengths? Mov
Disord. 27:1714–1717. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ho LW, Brown R, Maxwell M, Wyttenbach A
and Rubinsztein DC: Wild type Huntingtin reduces the cellular
toxicity of mutant Huntingtin in mammalian cell models of
Huntington's disease. J Med Genet. 38:450–452. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shirasaki DI, Greiner ER, Al-Ramahi I,
Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S,
Loo JA and Yang XW: Network organization of the huntingtin
proteomic interactome in mammalian brain. Neuron. 75:41–57.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nuzzo MT and Marino M:
Estrogen/Huntingtin: A novel pathway involved in neuroprotection.
Neural Regen Res. 11:402–403. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Leavitt BR, van Raamsdonk JM, Shehadeh J,
Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA and
Hayden MR: Wild-type huntingtin protects neurons from
excitotoxicity. J Neurochem. 96:1121–1129. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ismailoglu I, Chen Q, Popowski M, Yang L,
Gross SS and Brivanlou AH: Huntingtin protein is essential for
mitochondrial metabolism, bioenergetics and structure in murine
embryonic stem cells. Dev Biol. 391:230–240. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Guedes-Dias P, Pinho BR, Soares TR, de
Proença J, Duchen MR and Oliveira JM: Mitochondrial dynamics and
quality control in Huntington's disease. Neurobiol Dis. 90:51–57.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Brustovetsky N: Mutant huntingtin and
elusive defects in oxidative metabolism and mitochondrial calcium
handling. Mol Neurobiol. 53:2944–2953. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Grima JC, Daigle JG, Arbez N, Cunningham
KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer
JC, et al: Mutant Huntingtin disrupts the nuclear pore complex.
Neuron. 94:93–107.e6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zeitlin S, Liu JP, Chapman DL, Papaioannou
VE and Efstratiadis A: Increased apoptosis and early embryonic
lethality in mice nullizygous for the Huntington's disease gene
homologue. Nat Genet. 11:155–163. 1995.PubMed/NCBI View Article : Google Scholar
|
|
35
|
McKinstry SU, Karadeniz YB, Worthington
AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya
T, Dragatsis I, Zeitlin S, et al: Huntingtin is required for normal
excitatory synapse development in cortical and striatal circuits. J
Neurosci. 34:9455–9472. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mehler MF, Petronglo JR, Arteaga-Bracho
EE, Gulinello ME, Winchester ML, Pichamoorthy N, Young SK, DeJesus
CD, Ishtiaq H, Gokhan S and Molero AE: Loss-of-Huntingtin in medial
and lateral ganglionic lineages differentially disrupts regional
interneuron and projection neuron subtypes and promotes
Huntington's disease-associated behavioral, cellular, and
pathological Hallmarks. J Neurosci. 39:1892–1909. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jung R, Lee Y, Barker D, Correia K, Shin
B, Loupe J, Collins RL, Lucente D, Ruliera J, Gillis T, et al:
Mutations causing Lopes-Maciel-Rodan syndrome are huntingtin
hypomorphs. Hum Mol Genet. 30:135–148. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lackie RE, Maciejewski A, Ostapchenko VG,
Marques-Lopes J, Choy WY, Duennwald ML, Prado VF and Prado MAM: The
Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases.
Front Neurosci. 11(254)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Qi L, Zhang XD, Wu JC, Lin F, Wang J,
DiFiglia M and Qin ZH: The role of chaperone-mediated autophagy in
huntingtin degradation. PLoS One. 7(e46834)2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Keum JW, Shin A, Gillis T, Mysore JS, Abu
Elneel K, Lucente D, Hadzi T, Holmans P, Jones L, Orth M, et al:
The HTT CAG-Expansion mutation determines age at death but not
disease duration in huntington disease. Am J Hum Genet. 98:287–298.
2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Dulski J, Sulek A, Krygier M, Radziwonik W
and Slawek J: False-negative tests in Huntington's disease: A new
variant within primer hybridization site. Eur J Neurol.
28:2103–2105. 2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
De Luca A, Morella A, Consoli F, Fanelli
S, Thibert JR, Statt S, Latham GJ and Squitieri F: A Novel
Triplet-Primed PCR assay to detect the full range of trinucleotide
CAG repeats in the huntingtin gene (HTT). Int J Mol Sci.
22(1689)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Langfelder P, Gao F, Wang N, Howland D,
Kwak S, Vogt TF, Aaronson JS, Rosinski J, Coppola G, Horvath S and
Yang XW: MicroRNA signatures of endogenous Huntingtin CAG repeat
expansion in mice. PLoS One. 13(e0190550)2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Reed ER, Latourelle JC, Bockholt JH, Bregu
J, Smock J, Paulsen JS and Myers RH: PREDICT-HD CSF Ancillary Study
Investigators. MicroRNAs in CSF as prodromal biomarkers for
Huntington disease in the PREDICT-HD study. Neurology.
90:e264–e272. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Glidden AM, Luebbe EA, Elson MJ,
Goldenthal SB, Snyder CW, Zizzi CE, Dorsey ER and Heatwole CR:
Patient-reported impact of symptoms in Huntington disease:
PRISM-HD. Neurology. 94:e2045–e2053. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Paulsen JS, Miller AC, Hayes T and Shaw E:
Cognitive and behavioral changes in Huntington disease before
diagnosis. Handb Clin Neurol. 144:69–91. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Oosterloo M, de Greef BTA, Bijlsma EK,
Durr A, Tabrizi SJ, Estevez-Fraga C, de Die-Smulders CEM and Roos
RAC: Disease onset in Huntington's disease: When is the conversion?
Mov Disord Clin Pract. 8:352–360. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Squadrone S, Brizio P, Abete MC and Brusco
A: Trace elements profile in the blood of Huntington' disease
patients. J Trace Elem Med Biol. 57:18–20. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Caron NS, Wright GEB and Hayden MR:
Huntington Disease. 1998 Oct 23 (updated 2020 Jun 11). In:
GeneReviews® (Internet). Adam MP, Ardinger HH, Pagon RA,
Wallace SE, Bean LJH, Mirzaa G and Amemiya A (eds). University of
Washington, Seattle, WA, 1993-2021.
|
|
50
|
Shoulson I and Young AB: Milestones in
huntington disease. Mov Disord. 26:1127–1133. 2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Pringsheim T, Wiltshire K, Day L, Dykeman
J, Steeves T and Jette N: The incidence and prevalence of
Huntington's disease: A systematic review and meta-analysis. Mov
Disord. 27:1083–1091. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sobel SK and Cowan DB: Impact of genetic
testing for Huntington disease on the family system. Am J Med
Genet. 90:49–59. 2000.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Migliore S, Jankovic J and Squitieri F:
Genetic Counseling in Huntington's disease: Potential new
challenges on Horizon? Front Neurol. 10(453)2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
MacLeod R, Metcalfe A and Ferrer-Duch M: A
family systems approach to genetic counseling: Development of
narrative inter-ventions. J Genet Couns. 30:22–29. 2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Pierron L, Hennessy J, Tezenas du Montcel
S, Coarelli G, Heinzmann A, Schaerer E, Herson A, Petit E, Gargiulo
M and Durr A: Informing about genetic risk in families with
Huntington disease: Comparison of attitudes across two decades. Eur
J Hum Genet. 29:672–679. 2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Barker RA, Fujimaki M, Rogers P and
Rubinsztein DC: Huntingtin-lowering strategies for Huntington's
disease. Expert Opin Investig Drugs. 29:1125–1132. 2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Aziz NA, van der Burg JMM, Tabrizi SJ and
Landwehrmeyer GB: Overlap between age-at-onset and
disease-progression determinants in Huntington disease. Neurology.
90:e2099–e2106. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Tabrizi SJ, Ghosh R and Leavitt BR:
Huntingtin lowering strategies for disease modification in
Huntington's disease,. Neuron. 101:801–819. 2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Lane RM, Smith A, Baumann T, Gleichmann M,
Norris D, Bennett CF and Kordasiewicz H: Translating antisense
technology into a treatment for Huntington's disease. Methods Mol
Biol. 1780:497–523. 2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB,
Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J,
Rickards H, et al: Targeting Huntingtin expression in patients with
Huntington's disease. N Engl J Med. 380:2307–2316. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Harper SQ, Staber PD, He X, Eliason SL,
Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL and Davidson BL:
RNA interference improves motor and neuropathological abnormalities
in a Huntington's disease mouse model. Proc Natl Acad Sci USA.
102:5820–5825. 2005.PubMed/NCBI View Article : Google Scholar
|
|
62
|
DiFiglia M, Sena-Esteves M, Chase K, Sapp
E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, et
al: Therapeutic silencing of mutant huntingtin with siRNA
attenuates striatal and cortical neuropathology and behavioral
deficits. Proc Natl Acad Sci USA. 104:17204–17209. 2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Keiser MS, Kordasiewicz HB and McBride JL:
Gene suppression strategies for dominantly inherited
neurodegenerative diseases: Lessons from Huntington's disease and
spinocerebellar ataxia. Hum Mol Genet. 25(R1):R53–R64.
2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Yu D, Pendergraff H, Liu J, Kordasiewicz
HB, Cleveland DW, Swayze EE, Lima WF, Crooke ST, Prakash TP and
Corey DR: Single-stranded RNAs use RNAi to potently and
allele-selectively inhibit mutant huntingtin expression. Cell.
150:895–908. 2012.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Dufour BD, Smith CA, Clark RL, Walker TR
and McBride JL: Intrajugular vein delivery of AAV9-RNAi prevents
neuropathological changes and weight loss in Huntington's disease
mice. Mol Ther. 22:797–810. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Miniarikova J, Zanella I, Huseinovic A,
van der Zon T, Hanemaaijer E, Martier R, Koornneef A, Southwell AL,
Hayden MR, van Deventer SJ, et al: Design, Characterization, and
lead selection of therapeutic miRNAs targeting huntingtin for
development of gene therapy for Huntington's disease. Mol Ther
Nucleic Acids. 5(e297)2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Devi S, Kumar V, Singh SK, Dubey AK and
Kim JJ: Flavonoids: Potential candidates for the treatment of
neurodegenerative disorders. Biomedicines. 9(99)2021.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Huntington Study Group. Tetrabenazine as
antichorea therapy in Huntington disease: A randomized controlled
trial. Neurology. 66:366–372. 2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Arora A, Kumar S, Ali J and Baboota S:
Intranasal delivery of tetrabenazine nanoemulsion via olfactory
region for better treatment of hyperkinetic movement associated
with Huntington's disease: Pharmacokinetic and brain delivery
study. Chem Phys Lipids. 230(104917)2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Catanesi M, Caioni G, Castelli V,
Benedetti E, d'Angelo M and Cimini A: Benefits under the Sea: The
role of marine compounds in neurodegenerative disorders. Mar Drugs.
19(24)2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Jabłońska M, Grzelakowska K, Wiśniewski B,
Mazur E, Leis K and Gałązka P: Pridopidine in the treatment of
Huntington's disease. Rev Neurosci. 31:441–451. 2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Squitieri F, Di Pardo A, Favellato M,
Amico E, Maglione V and Frati L: Pridopidine, a dopamine
stabilizer, improves motor performance and shows neuroprotective
effects in Huntington disease R6/2 mouse model. J Cell Mol Med.
19:2540–2548. 2015.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Garcia-Miralles M, Geva M, Tan JY, Yusof
NABM, Cha Y, Kusko R, Tan LJ, Xu X, Grossman I, Orbach A, et al:
Early pridopidine treatment improves behavioral and transcriptional
deficits in YAC128 Huntington disease mice. JCI insight.
2(e95665)2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Bartl S, Oueslati A, Southwell AL, Siddu
A, Parth M, David LS, Maxan A, Salhat N, Burkert M, Mairhofer A, et
al: Inhibiting cellular uptake of mutant huntingtin using a
monoclonal antibody: Implications for the treatment of Huntington's
disease. Neurobiol Dis. 141(104943)2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Connor B: Concise review: The use of stem
cells for understanding and treating Huntington's disease. Stem
Cells. 36:146–160. 2018.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Fritz NE, Rao AK, Kegelmeyer D, Kloos A,
Busse M, Hartel L, Carrier J and Quinn L: Physical therapy and
exercise interventions in Huntington's disease: A mixed methods
systematic review. J Huntingtons Dis. 6:217–235. 2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Roos RA: Huntington's disease: A clinical
review. Orphanet J Rare Dis. 5(40)2010.PubMed/NCBI View Article : Google Scholar
|