1. Introduction
Mitochondria serve critical biological roles in the
cell, including the production and control of reactive oxygen
species (ROS), preservation of Ca2+ ion homeostasis, and
modulation of programmed cell death (1,2). As an
essential organelle, mitochondria serve a role in the generation of
fatty acids and integrate the cell signaling circuitry that
modulates cell survival, immune response, and autophagy (3,4).
Therefore, the dysfunction of mitochondria can trigger damage
(5,6).
As the energy generator, the genome of mitochondria,
namely mitochondrial DNA (mtDNA), encodes 37 key genes and 13
mitochondrial proteins, two RNAs and 22 transfer RNAs (3,7,8). In
response to cellular stress that triggers unbalanced intracellular
processes such as respiration, energy production in the form of
ATP, or apoptosis, mtDNA fragments are released as cell-free mtDNA
(cf-mtDNA) in the bloodstream (9-11).
cf-mtDNA is smaller compared with nuclear cfDNA. Moreover, cf-mtDNA
is present in large amounts in patients with hepatocellular and
prostate cancer (9,12).
Mitochondria interact with organelles, such as
endoplasmic reticulum, lysosomes, cytoskeleton, peroxisomes, and
nucleus, to accomplish their bioenergetics, metabolism, and
apoptosis roles (11,13). These processes were predicted to be
fundamentally active and are not controlled by other reactions.
Previous work has reported the arrangement of indiscriminate
mitochondrial protein import (4).
This activity represents the health of mitochondria as well as the
functional condition (14). The
function of mitochondria is associated with transcription in the
nucleus in response to intrinsic and extrinsic signals, such as
modulation of the intracellular reduction-oxidation state, nutrient
deprivation and exercise (15,16).
A previous lineage tracer study reported that
haplogroup U5 is the most ancient haplogroup in Europe (17). Many studies categorize mtDNA in
haplogroups, which is useful to trace maternal phylogenetic
lineages, and is synergistically communicated from mitochondrial
genome. The U5b2c1 haplogroup is found uncommon in modern
populations (17-19).
The phenotypes of the disease are associated with specific sequence
mutations in mtDNA variations, for example, mitochondrial
encephalopathy lactic acidosis with stroke-like episodes) syndrome
Clinical phenotypes are correlated with human mtDNA pathogenic
mutation, e.g., in patients with Kearns Sayre syndrome). Therefore,
a mismatch between the nuclear and mitochondrial genome may affect
mitochondria quality and leads to cellular problems, such as
respiratory dysfunction, DNA damage (20-22).
The present review aimed to assess the molecular
mechanism and potential roles of mitochondria in neuro-aging,
including the importance of evaluating the health status of mtDNA
via mitochondrial dynamics.
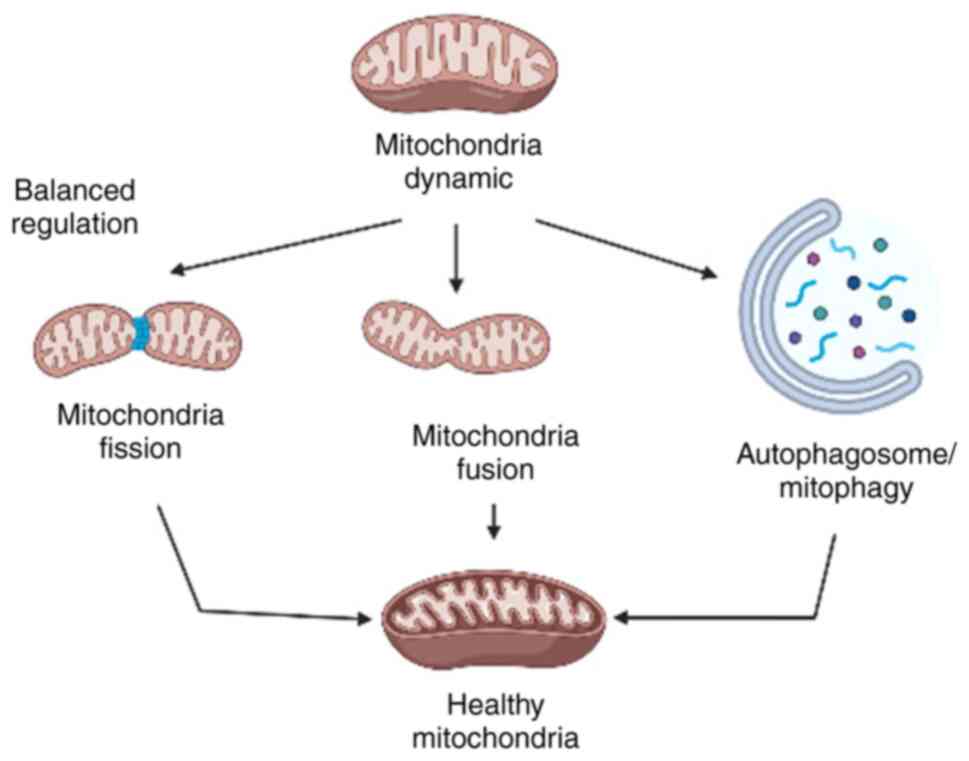
2. Balance quality control role of
mitochondria leads to healthy mitochondrial dynamics
The quality control process of mitochondria, which
includes fission and fusion, mitophagy, transport and biogenesis,
sustains its balance and supports cellular health (23). Specifically, the normal condition of
mitochondria, defined as mitochondrial dynamics, involves changes
of morphology due to fission and fusion events (24). During traumatic conditions, such as
lack of nutrition, two mutated genes may merge and allow functional
complementation by RNA, resulting in new mitochondria and
maintaining its respiratory function (25). The basic concept of balanced
mitochondrial dynamics that leads to healthy mitochondria is
presented in Fig. 1.
By contrast, the fission process triggers the
breakdown of mitochondria into smaller fragments to facilitate
transport and autophagy (13).
Fission often induces dissociation of damaged components, thus, the
ability of mitophagy is impacted by impaired fission (26). Mitophagy is key to remove any
malfunctioning or unneeded mitochondria and direct them to
autophagosomes to sustain quality control (27).
To manage organelle and protein turnover within
cells, autophagy and apoptosis perform mitochondria quality control
(5,17). Autophagy works by recycling
selective intracellular organelles, while apoptosis works by
removing damaged cells. In certain conditions, e.g., in severe cell
damage, autophagy induces apoptosis or necrosis by degrading the
cytoplasm excessively. There is a unique cellular response which
induces autophagy and the mechanisms of cell death that leads to
normal removal of dead cells, as well as immune recognition of
antigens (28). Mitochondria serve
an essential role in the connection between autophagy and apoptosis
that influences pathological conditions (29).
Mitochondria manage damaged proteins or remove
abnormal organelles to maintain their function (27). Impairment of quality control
decreases of the balance of normal cellular function and lead to
decreased survival and general cell health associated with aging
(30). Thus, mitochondrial
impairment and dysregulation are associated with neurodegenerative
disorders (Alzheimer's disease, Parkinson's disease, Huntington's
disease, and amyotrophic lateral sclerosis), metabolic syndromes,
and cancer (5).
In addition, mitochondria fission increases the
number of organelles to activate biogenesis and remove injured
organelles for autophagic degradation (31). The balance between fusion and
fission regulates distribution of mtDNA, mitochondrial biogenesis
and proteins (11). Abnormal
function of chaperones and proteases may lead to accumulation of
unfolded/misfolded/damaged proteins/protein aggregates and orphaned
subunits (7,4,27).
Quality control of impaired mitochondria is a therapeutic approach
in many types of neurodegenerative disease, e.g., Parkinson's
disease, Huntington's disease, and Alzheimer's disease. It is
hypothesized that these neurodegenerative diseases associated with
mitochondrial genetic improvement are also associated with
mitochondrial disease and disorder (28,32).
3. Mitochondria serve an essential role in
response to neuro-aging
In relation to cellular health, mitochondria are
actively studied; mitochondrial impairment due to cellular
senescence is associated with certain types of neurodegenerative
diseases (33,34). Mitochondrial-associated disorder or
mitochondrial dysfunctions such as neurodegenerative disorders,
neurometabolic diseases, neuroaging, have a prevalence of 1:2,000
individuals (17,13). Mitochondrial-associated disorders
mostly are caused by the mutations of genes that alter mtDNA
replication and transcription and mitochondrial mRNA translation,
thus decreasing the mitochondrial oxidative phosphorylation
(OXPHOS). The inhibition of OXPHOS process may also be caused by
dysfunction of respiratory chain enzyme complexes and ATP synthase
in the mitochondrial inner membrane folds. This dysfunction leads
to cell-specific stress responses and health problems, e.g.,
neuroaging or neurodegenerative disorders (10).
Copy number of mtDNA (mtDNA-cn) in mitochondria
genome represents cellular health. cn can be measured in peripheral
blood mononuclear cells (7,35). Studies have shown that mtDNA is
liberated in small amounts in the blood under cellular stress and
can be detected in plasma in the form of circulating cf-mtDNA
(ccf-mtDNA) (6,9). Furthermore, imbalance between
production and accumulation of ROS in cells and tissue may lead to
the movement of mtDNA into the cytoplasm and the extracellular
region. This is triggered by long-term neuroendocrine,
inflammatory, oxidative and metabolic stress and may result in
mitochondrial dysfunction and mtDNA damage (6,9). The
effect of unbalanced mitochondrial dynamics on mitochondria is
presented in Fig. 2.
cfDNA derived from mtDNA has been widely
investigated in cancer and inflammatory, cardiovascular, and
metabolic disease (10,11,36).
Therefore, mitochondrial health is a novel approach to discover
potential indicators for neurological and brain health in
conditions such as depressive disorders, Alzheimer's disease,
dementia and Parkinson's disease (35,37).
Levels of ccf-mtDNA and cellular mtDNA-cn may be associated with
oxidative stress and cellular senescence. mtDNA is vulnerable to
oxidative stress due to lack of protective histones and limited DNA
repair mechanisms. Glutathione peroxidase, an antioxidant enzyme,
is required to protect the mitochondria and prevent dysfunction
(38).
Studies have shown that mtDNA mutations are
positively related with aging, depression and Parkinson's and
Alzheimer's disease (11,15). Evidence has shown that dysregulated
mitochondrial dynamics and mutations due to mtDNA replication
induce aging and increased mtDNA mutation rates is correlated with
the aging rate (11,15). High levels of mtDNA deletion are
found in the substantia nigra neurons of aging patients with
Parkinson's disease. mtDNA deletion cause human disease and their
accumulation play a role in the aging process (39). Parkinson's disease is a
neurodegenerative disease characterized by a depletion of dopamine
neurons in the basal ganglia in the midbrain and the accumulation
of synuclein (13,30).
Studies on Parkinson's disease have indicated a
defect in Parkin RBR E3 ubiquitin-protein ligase and PTEN-induced
kinase 1 (PINK1) (30,38). An autosomal recessive mutation at
two genes (PARK2 and PARK6) that encode outer mitochondrial
membrane kinase PINK1 and the cytosolic Parkin E3 ubiquitin-protein
ligase, lead to the reduction of mitochondrial damage (30,38).
Impairment of these processes leads to loss of muscle and
dopaminergic neurons and affects male infertility in a way that can
be fixed by inducing mitochondrial fission (40). As a novel biomarker of depression,
ccf-mtDNA levels exhibit a positive correlation with inflammatory
symptoms of depression (41). A
recent cohort study of patients with depressive disorder (n=281;
36% with a personality disorder and 13% with bipolar; 93% receiving
mono- or multi-psychotropic drugs), demonstrated a significant
difference in mean ccf-mtDNA levels between patients with a current
(n=236) or remitted depressive episode (n=45) and healthy
participants (n=49); treatment using mood stabilizers, such as
lamotrigine, valproic acid or lithium, was associated with
decreased ccf-mtDNA proportion (41). The aforementioned study hypothesized
that mtDNA is recognised as a damage-associated molecular pattern
(DAMP), activating the innate immune response primarily by
attaching to the toll-like receptor 9. The aforementioned study
suggested that ccf-mtDNA may be differentially regulated in
different subtypes of depression. ccf-mtDNA in inflammatory
depression subtype is correlated with worse treatment response to
conventional selective serotonin reuptake inhibitors and a more
prospective anti-inflammatory agent, such as omega-3 fatty acids
(41). The mitochondrial studies in
patients with neurodisorders are summarized in Table I.
 | Table IMitochondrial biomarkers for
neuro-aging prediction. |
Table I
Mitochondrial biomarkers for
neuro-aging prediction.
| First author,
year | Mitochondrial
biomarker | Clinical
results | (Refs.) |
|---|
| Park et al,
2022 | Peripheral and CSF
ccf-mtDNA of neuropsychiatric patients | No significant
difference in levels of peripheral ccf- mtDNA in neuropsychiatric
studies between cases and controls. CSF ccf-mtDNA levels in non-
psychiatric neurological disease decreased compared with
controls | (1) |
| Peng et al,
2019 | CSF cf-mtDNA of
patients with NMDAR | Significantly
higher levels of CSF cf-mtDNA and inflammation-associated cytokines
in patients with NMDAR | (2) |
| Lindqvist et
al, 2018 | ccf-mtDNA of
patients with MDD | Significantly
elevated levels of ccf-mtDNA in patients with MDD | (7) |
| Newell et
al, 2018 | Plasma mtDNA of
patients with mitochondrial disease | Full mitochondrial
genome presents in the cf plasma fraction of human blood | (36) |
| Castellazzi et
al, 2019 | Autophagy (ATG5
protein) and mitophagy marker (Parkin protein) in patients with AD
and MCI | Patients with AD
and MCI showed significantly decreased circulating levels of both
ATG5 and Parkin compared with healthy controls | (38) |
| Kageyama et
al, 2018 | Plasma mtDNA of
patients with MDD, BD and SZ | Patients with MDD
and BD showed significantly lower plasma mtDNA levels than
controls. Plasma mtDNA is associated with cytokines; GM- CSF, IL-2,
and IL-4 in patients with MDD mtDNA levels were lower in the
depressive state than in the remission state in patients with
MDD | (39) |
| Trumpff et
al, 2019 | Serum ccf-mtDNA of
healthy midlife adults exposed to acute psychological
challenge | Acute psychological
stress increased ccf-mtDNA levels. Neuroendocrine signaling
triggered mtDNA extrusion in living cells | (62) |
| Silzer et
al, 2019 | Plasma ccf-mtDNA,
cf-mtDNA, mtDNA copy number in patients with T2DM and AD | Cf-mtDNA levels are
higher in individuals with T2DM but not significantly in those with
cognitive impairment compared with controls | (63) |
Alzheimer's disease is associated with heteroplasmic
mtDNA mutations. This disease is characterized by the formation of
amyloid-β (Aβ) plaques in the extracellular space, which exhibits
substantial neuronal loss (4).
Accumulation of Aβ plaques increases oxidative damage in neurons,
is associated with the accumulation of reactive oxygen species in
the mitochondrial (42), and
induces the severity of mtDNA damage. Eventually, this condition
steers to the rise of oxidized nucleic acid in mtDNA (43).
4. Mitochondrial dynamics as a key factor in
predicting an early neuro-aging
Mitochondrial dynamics are critical contributors in
predicting the development of neuro-aging (44). Mitochondrial dynamics influence the
self-renewal and differentiation of neuron in cell metabolism
(13,44). In neurons of neonates, small
mitochondria fission is observed, which size increases during
maturity process. By contrast, the developing brain exhibits more
fused and fragmented mitochondria (33,43).
In neurogenesis, mitochondrial dynamics is associated with cell
cycle; there is increased escalated fusion during G1/S phase,
followed by fission during G2 and mitosis (5,45).
Mitochondrial fission occurs during neural stem progenitor cell
(NSPC) mitosis, which exhibits dichotomic behaviour of the daughter
cells: Those destined to remain NSPCs display high levels of
mitochondrial fusion, whereas prospective neuronal cells maintain
higher levels of mitochondrial fission (24,46).
With aging, mitochondria will accumulate oxidative
damage with reduced reparative ability leading to impairment and
dysfunction (26). Escape of
electrons during the electron transport cascade in OXPHOS may
generate oxidative stress and disrupt the cellular metabolic and
signalling routes (16).
Mitochondrial fission and fusion are associated with mitochondrial
activity. These activities, (such as programmed cell death
regulation, haem complexes biosynthesis, calcium signalling, fatty
acids oxidation, and, a platform for signal transduction in the
innate immune response, are modulated in mitochondria cristae, in
which OXPHOS molecular effectors and electron transport chain
components are concentrated (45).
Mitochondrial damage may cause cognitive impairment, increased
aggregation and neural disorders such as Alzheimer's disease,
Parkinson's disease, Huntington's disease, and ischemic stroke
(46).
In neurogenesis, mitochondria activity plays an
essential role in the amplification and differentiation of
neurogenic precursors (47). During
mitosis, mitochondria of the neural stem cells (NSCs) are present
mainly in the form of fragmented mitochondria, and during the
G0/early G1 phase (1 h after mitosis) these fragmented mitochondria
undergo differentiation and progressive fusion in neurogenic
precursors or form immature neuron (44). It is hypothesized that the
association between mitochondria morphology, dynamics and function
serve an essential role in neurogenesis as a response to
neuro-aging (48,49). OXPHOS in human neurogenesis is to
facilitate the renewal of NSC and the acquisition of neuronal fate
by generating ATP in cells (37).
More than 98% of essential proteins for mitochondrial function are
encoded in the nucleus, translated in the cytoplasm based on the
presence of encoded mitochondrial targeting sequences. These
proteins are imported to specific sub-compartments to stimulate the
physiological activity of mitochondrial enzymes, such as
translocases, proteinase and chaperones (50). This process is the most important
character of mitochondrial dynamics, by which cellular homeostasis
is coordinated via communication between the mitochondria and the
nucleus, thus modulating adaptive responses to stress (31,45).
Mitochondrial dynamics determines the distribution
and maintenance of mtDNA in the mitochondrial network (50). Studies have shown an association
between mtDNA, dynamin related protein-1 and endoplasmic reticulum
in mitochondrial replication and fission, which lead to equal
distribution of mtDNA in mitochondria and cells (7,43).
Lack of fission and fusion in mitochondria lead to genetic
dysfunction such as severe or multiple deletion, increased levels
and unequal distribution of mtDNA (51). Impairment of mitochondrial fission
induces disorganization of inner mitochondrial structure membranes,
such as cristae junctions, which maintain proper internal membrane
compartmentalization; loss of these junctions leads to clustering
and mis-segregation of mtDNA nucleoids (48,52).
When disorganization of the mitochondrial structure occurs, the
ability of mtDNA to repair cellular damage will decrease, leading
to the false protection to neuro-aging process (16).
During aging, the aperture of the mitochondrial
permeability transition pore (mPTP) is associated with
depolarization of mitochondria and OXPHOS uncoupling. Smaller
molecules (#x003C;1,500 Da) enter mitochondria via the mPTP
(29). Under physiological
conditions, the mPTP serves as an efflux channel for calcium ions.
mPTP is activated by ROS, which causes the release of cytochrome-C
oxidase and the initiation of the caspase-9 cascade, which
activates the apoptotic pathway (22,33).
The nod-like receptor pyrin domain 3 (NLRP3) inflammasome may also
be activated as a result of mPTP activation and subsequent ROS
generation. This is key because neuronal loss in nigrostriatal
neurons is caused by NLRP3 activation in brain microglia and
astrocytes; this activation is a fundamental mechanism by which
Parkinson's disease and other illnesses display motor impairments
and cognitive abnormalities (30,32).
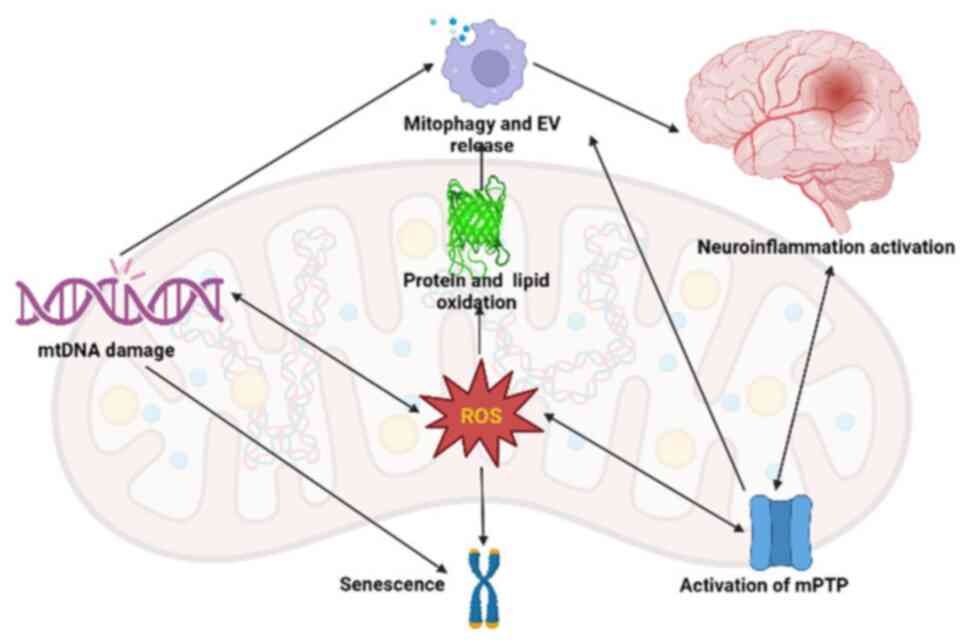
Beside the balance between fission and fusion of
mitochondria, another option to maintain healthy mitochondria is to
release toxic substances, including oxidized cardiolipin, protein
and mtDNA, into extracellular vesicle (EVs), which are subsequently
degraded. EV generation rises with aging and is correlated with
mtDNA release and pro-inflammatory cytokine production (48). The components of EV, such as mtDNA,
ROS and cardiolipin, serve as DAMPs, disrupt nearby molecular
patterns and trigger inflammatory responses that lead to
neuro-aging (Fig. 3) (50,52).
Cardiolipin and mitochondrial dysfunction are required for NLRP3
activation. Caspases 1 and 2 are activated as a result of NLRP3
induction, cleaving Parkin to stop mitophagy (53). When fission and fusion are no longer
possible, oxidized macromolecules accumulate and activate IL-1 and
IL-18 to cause pyroptosis, an inflammation-mediated cell death
(42). Following cell death, mtDNA
and ROS are released and interact with other inflammasomes to
exacerbate the inflammatory response (53).
Any changes to mtDNA that codes for cytochrome C
oxidase or direct protein oxidation of the enzyme may cause early T
cell death and loss of function, which may explain immune system
impairment in the elderly (25,27).
Microglia in the brain and immuno-senescence of T cells have both
been connected to mitochondrial dysfunction. This could lead to
aberrant activation in response to damage. However, mitochondrial
malfunction in microglia inhibits the ability of T cells and
microglia to adopt a neuroprotective role (27,53).
It is hypothesized that aging-associated neuroinflammatory
processes in microglia alter the synaptic plasticity of the brain
and impair memory by causing downstream changes (27,53).
Astrocytic activation is stimulated by microglial activation and
serves a crucial part in both neuroinflammation and the aging of
the brain (25,53).
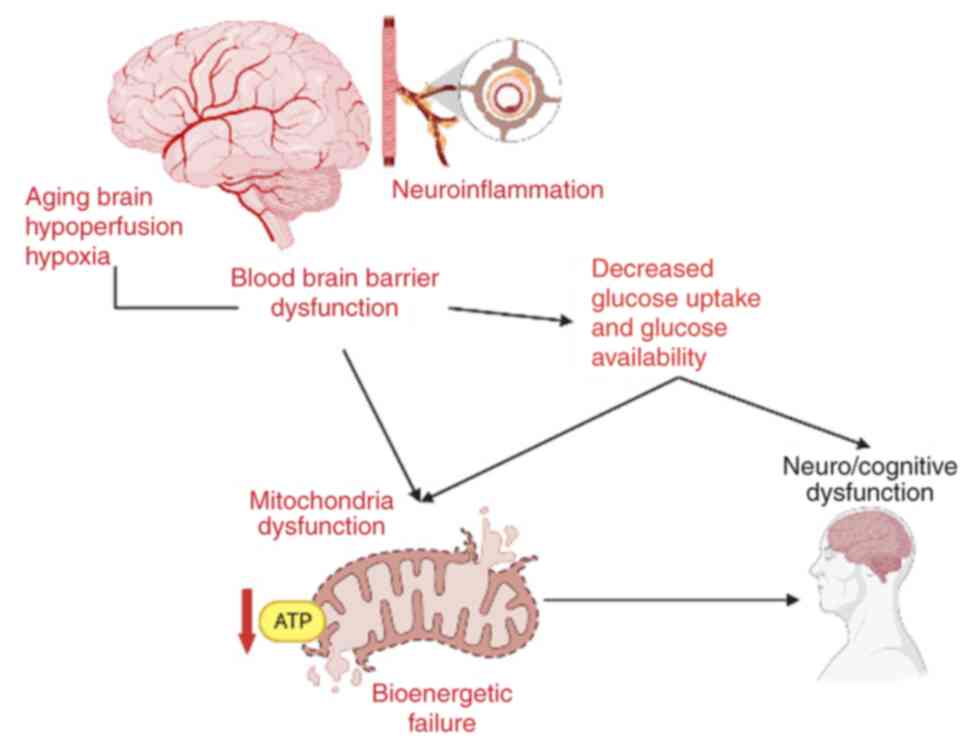
Due to limited intracellular glycogen stores in the
brain and the high energy needs of neurons, age-associated
hypoperfusion has notable effects on mitochondrial energy
metabolism and cognitive functioning (39,53).
The association between aging brain, hypoperfusion, hypoxia (due to
neuroinflammation) and BBB dysfunction that leads to decreased
glucose uptake and mitochondrial dysfunction, is depicted in
Fig. 4. Age-associated cognitive
impairment is linked to hypometabolism, which is characterized by
decreased oxidative respiration efficiency, ATP generation and
expression of genes implicated in mitochondrial biogenesis,
oxidative respiration and dysregulation (42,46).
Using directly reprogrammed neurons (iN) from elderly donors to
study mitochondrial function in aging shows decreased energy output
and significant downregulation of genes involved in the electron
transport chain complexes I, III, IV and V, accounting for 70% of
all mitochondrial genes when compared with iN from young donors;
proteins encoded by genes for mitochondrial phosphorylation
decreased along with mRNA and protein expression (53).
To confirm that abnormality of mitochondrial
dynamics promote ageing in neurons, physiological factors in
cellular functions and processes related to mitochondrial fitness
must be maintained (45). The
depletion of mitofusin-2 (MFN-2) a fusion GTPase, in mitochondria
in human cells stimulates mitochondrial dysfunction, characterized
by decreased ATP synthesis, increased proton leak, reduced
mitochondrial membrane potential and elevated production of ROS
(38). MFN-2 modulates mitochondria
cristae, which are associated with decreased mitophagy and impact
mitochondria quality control ability (54). Impairments in mitochondrial quality
control are associated with impaired neuronal development,
plasticity and function, and therefore are involved in several
neurodegenerative diseases, such as Parkinson's, Alzheimer's and
Huntington's disease (55).
To maintain a healthy mitochondrial population, the
removal of damaged mitochondria via autophagy and mitophagy and
stimulation of mitochondrial biogenesis must be controlled
(48,56). During aging, autophagy and mitophagy
are commonly impaired, resulting in alteration of several types of
protein, e.g., PINK-1 (37,55). The impairment of mitophagy is
related to aging and age-associated disease (51,52).
Changes in mitochondrial fusion or fission proteins trigger
intrinsic mitochondrial defects that result in a decrease in
mitochondrial activity and an increase in mitochondrial damage
(47,57). Impaired autophagy and/or mitophagy
and decreased elimination of damaged mitochondria are influenced by
unbalanced mitochondrial dynamics (26,58).
Abnormal mitochondrial dynamics may induce damaged
mitochondria (59). Mitochondrial
dysfunction and decreased autophagy and/or mitophagy are promote
dysregulation of mitochondrial dynamics during aging (60,61).
Dysfunction in mitochondrial dynamics is correlated with
age-related disease and health span, particularly in humans
(46). Neuro-aging is associated
with the dysregulation of mitochondrial dynamics, triggered by
age-related disorders via various mechanisms and signalling routes
(54,62). Accumulation of damaged mitochondria
that cannot be processed by autophagy or mitophagy may result in
the dysfunction of mtDNA, which is associated with
neurodegeneration (55,63).
This review is in the framework of the first
author's dissertation project which concerns to finding an early
detection of mild cognitive impairment and its progression in
elderly patients. In this article we did not specify the technical
issue as organelles separation and preservations.
5. Conclusion
Previous studies (27,54,55,52,63)
have investigated the role of mtDNA to find potential early markers
to predict and evaluate neurodegenerative processes related to
aging. Approaches to evaluate healthy mitochondria include
mitochondria dynamics, which is associated with mitochondria
quality control. It is hypothesized that mitochondrial functions
(fusion, fission, mitophagy and DNA repair) should be assessed as
complete indicators to determine health status of mitochondria in
aging. However, research on the role of mtDNA to find an early
marker to predict neurodegeneration associated with aging is
needed.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
DM and MPS were responsible for the conception and
design of the study. MPS performed the literature review and wrote
the manuscript. JL, MPS and EH were responsible for reviewing and
revising the manuscript. Data authentication is not applicable. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Park SS, Jeong H and Andreazza AC:
Circulating cell-free mitochondrial DNA in brain health and
disease: A systematic review and meta-analysis. World J Biol
Psychiatry. 23:87–102. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Peng Y, Zheng D, Zhang X, Pan S, Ji T,
Zhang J, Shen HY and Wang HH: Cell-free mitochondrial DNA in the
CSF: A potential prognostic biomarker of Anti-NMDAR encephalitis.
Front Immunol. 10(103)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chang CY, Liang MZ and Chen L: Current
progress of mitochondrial transplantation that promotes neuronal
regeneration. Transl Neurodegener. 8(17)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Moya GE, Rivera PD and Dittenhafer-Reed
KE: Evidence for the role of mitochondrial DNA release in the
inflammatory response in neurological disorders. Int J Mol Sci.
22(7030)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cai Q and Tammineni P: Alterations in
mitochondrial quality control in Alzheimer's disease. Front Cell
Neurosci. 10(24)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Valenti D, Braidy N, De Rasmo D, Signorile
A, Rossi L, Atanasov AG, Volpicella M, Henrion-Caude A, Nabavi SM
and Vacca RA: Mitochondria as pharmacological targets in Down
syndrome. Free Radic Biol Med. 114:69–83. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lindqvist D, Wolkowitz OM, Picard M,
Ohlsson L, Bersani FS, Fernström J, Westrin Å, Hough CM, Lin J,
Reus VI, et al: Circulating cell-free mitochondrial DNA, but not
leukocyte mitochondrial DNA copy number, is elevated in major
depressive disorder. Neuropsychopharmacol. 43:1557–1564.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Aryaman J, Bowles C, Jones NS and Johnston
IG: Supplemental Material for Aryaman et al., 2019.
Figshare; 2019 [cited 2022 Sep 11]. p. 1311221 Bytes. Available
from: https://gsajournals.figshare.com/articles/Supplemental_Material_for_Aryaman_et_al_2019/8343830.
|
|
9
|
An Q, Hu Y, Li Q, Chen X, Huang J,
Pellegrini M, Zhou XJ, Rettig M and Fan G: The size of cell-free
mitochondrial DNA in blood is inversely correlated with tumor
burden in cancer patients. Precision Clinical Medicine. 2:131–139.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu B, Ni H, Li J, Zhuang X, Zhang J, Qi Z,
Chen Q, Wen Z, Shi H, Luo X and Jin B: The impact of circulating
mitochondrial DNA on cardiomyocyte apoptosis and myocardial injury
after TLR4 activation in experimental autoimmune myocarditis. Cell
Physiol Biochem. 42:713–728. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yan C, Duanmu X, Zeng L, Liu B and Song Z:
Mitochondrial DNA: Distribution, mutations, and elimination. Cells.
8(379)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gambardella S, Limanaqi F, Ferese R,
Biagioni F, Campopiano R, Centonze D and Fornai F: ccf-mtDNA as a
potential link between the brain and immune system in
neuro-immunological disorders. Front Immunol.
10(1064)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gao J, Wang L, Liu J, Xie F, Su B and Wang
X: Abnormalities of mitochondrial dynamics in neurodegenerative
diseases. Antioxidants. 6(25)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang AS and Dreesen O: Biomarkers of
cellular senescence and skin aging. Front Genet.
9(247)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Pugazhenthi S: Metabolic syndrome and the
cellular phase of Alzheimer's disease. Prog Mol Biol Transl Sci.
146:243–258. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hood S and Amir S: The aging clock:
Circadian rhythms and later life. J Clin Invest. 127:437–446.
2017.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Malyarchuk B, Derenko M, Grzybowski T,
Perkova M, Rogalla U, Vanecek T and Tsybovsky I: The peopling of
Europe from the mitochondrial haplogroup U5 perspective. PLoS One.
5(e10285)2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Meiliana A, Dewi NM and Wijaya A:
Mitochondria in health and disease. Indones Biomed J. 11:1–15.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Richards M, Macaulay V, Hickey E, Vega E,
Sykes B, Guida V, Rengo C, Sellitto D, Cruciani F, Kivisild T, et
al: Tracing European founder lineages in the Near Eastern mtDNA
pool. Am J Hum Genet. 67:1251–1276. 2000.PubMed/NCBI
|
|
20
|
de Goede P, Wefers J, Brombacher EC,
Schrauwen P and Kalsbeek A: Circadian rhythms in mitochondrial
respiration. J Mol Endocrinol. 60:R115–R130. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Oliver D and Reddy P: Dynamics of
dynamin-related protein 1 in Alzheimer's disease and other
neurodegenerative diseases. Cells. 8(961)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dunham-Snary KJ and Ballinger SW:
GENETICS. Mitochondrial-nuclear DNA mismatch matters. Science.
349:1449–1450. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hummel EM, Hessas E, Müller S, Beiter T,
Fisch M, Eibl A, Wolf OT, Giebel B, Platen P, Kumsta R and Moser
DA: Cell-free DNA release under psychosocial and physical stress
conditions. Transl Psychiatry. 8(236)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chakravorty A, Jetto CT and Manjithaya R:
Dysfunctional mitochondria and mitophagy as drivers of Alzheimer's
disease pathogenesis. Front Aging Neurosci. 11(311)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sebastián D, Palacín M and Zorzano A:
Mitochondrial dynamics: Coupling mitochondrial fitness with healthy
aging. Trends Mol Med. 23:201–215. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bhatia D, Chung KP, Nakahira K, Patino E,
Rice MC, Torres LK, Muthukumar T, Choi AM, Akchurin OM and Choi ME:
Mitophagy-dependent macrophage reprogramming protects against
kidney fibrosis. JCI Insight. 4(e132826)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Schrepfer E and Scorrano L: Mitofusins,
from mitochondria to metabolism. Mol Cell. 61:683–694.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gómez-Serrano M, Camafeita E, Loureiro M
and Peral B: Mitoproteomics: Tackling mitochondrial dysfunction in
human disease. Oxid Med Cell Longev. 2018(1435934)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Leung K, Chakraborty K, Saminathan A and
Krishnan Y: A DNA nanomachine chemically resolves lysosomes in live
cells. Nature Nanotech. 14:176–183. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang CW, Hang L, Yao TP and Lim KL:
Parkin regulation and neurodegenerative disorders. Front Aging
Neurosci. 7(248)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nicolas E, Tricarico R, Savage M, Golemis
EA and Hall MJ: Disease-associated genetic variation in human
mitochondrial protein import. Am J Hum Genet. 104:784–801.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Truban D, Hou X, Caulfield TR, Fiesel FC
and Springer W: PINK1, Parkin, and mitochondrial quality control:
What can we learn about Parkinson's disease pathobiology? J
Parkinsons Dis. 7:13–29. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Misgeld T and Schwarz TL: Mitostasis in
neurons: Maintaining mitochondria in an extended cellular
architecture. Neuron. 96:651–666. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Clark A and Mach N: The crosstalk between
the gut microbiota and mitochondria during exercise. Front Physiol.
8(319)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nicholas D, Proctor EA, Raval FM, Ip BC,
Habib C, Ritou E, Grammatopoulos TN, Steenkamp D, Dooms H, Apovian
CM, et al: Advances in the quantification of mitochondrial function
in primary human immune cells through extracellular flux analysis.
PLoS One. 12(e0170975)2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Newell C, Hume S, Greenway SC, Podemski L,
Shearer J and Khan A: Plasma-derived cell-free mitochondrial DNA: A
novel non-invasive methodology to identify mitochondrial DNA
haplogroups in humans. Mol Genet Metab. 125:332–337.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Martin LJ: Biology of mitochondria in
neurodegenerative diseases. Prog Mol Biol Transl Sci. 107:355–415.
2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Castellazzi M, Patergnani S, Donadio M,
Giorgi C, Bonora M, Bosi C, Brombo G, Pugliatti M, Seripa D,
Zuliani G and Pinton P: Autophagy and mitophagy biomarkers are
reduced in sera of patients with Alzheimer's disease and mild
cognitive impairment. Sci Rep. 9(20009)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kageyama Y, Kasahara T, Kato M, Sakai S,
Deguchi Y, Tani M, Kuroda K, Hattori K, Yoshida S, Goto Y, et al:
The relationship between circulating mitochondrial DNA and
inflammatory cytokines in patients with major depression. J Affect
Disord. 233:15–20. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jin SM and Youle RJ: PINK1- and
Parkin-mediated mitophagy at a glance. J Cell Sci. 125:795–799.
2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fernström J, Ohlsson L, Asp M, Lavant E,
Holck A, Grudet C, Westrin Å and Lindqvist D: Plasma circulating
cell-free mitochondrial DNA in depressive disorders. PLoS One.
16(e0259591)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cai Q and Jeong YY: Mitophagy in
Alzheimer's disease and other age-related neurodegenerative
diseases. Cells. 9(150)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wang W, Zhao F, Ma X, Perry G and Zhu X:
Mitochondria dysfunction in the pathogenesis of Alzheimer's
disease: Recent advances. Mol Neurodegeneration.
15(30)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Iwata R and Vanderhaeghen P: Regulatory
roles of mitochondria and metabolism in neurogenesis. Curr Opin
Neurobiol. 69:231–240. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Blagov AV, Grechko AV, Nikiforov NG,
Borisov EE, Sadykhov NK and Orekhov AN: Role of impaired
mitochondrial dynamics processes in the pathogenesis of Alzheimer's
disease. Int J Mol Sci. 23(6954)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Apaijai N, Sriwichaiin S, Phrommintikul A,
Jaiwongkam T, Kerdphoo S, Chansirikarnjana S, Thongmung N,
Mahantassanapong U, Vathesatogkit P, Kitiyakara C, et al: Cognitive
impairment is associated with mitochondrial dysfunction in
peripheral blood mononuclear cells of elderly population. Sci Rep.
10(21400)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Starkov AA: An update on the role of
mitochondrial α-ketoglutarate dehydrogenase in oxidative stress.
Mol Cell Neurosci. 55:13–16. 2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Shamsi TN, Athar T, Parveen R and Fatima
S: A review on protein misfolding, aggregation and strategies to
prevent related ailments. Int J Biol Macromol. 105:993–1000.
2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Stephan BCM, Hunter S, Harris D, Llewellyn
DJ, Siervo M, Matthews FE and Brayne C: The neuropathological
profile of mild cognitive impairment (MCI): A systematic review.
Mol Psychiatry. 17:1056–1076. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ding WX and Yin XM: Mitophagy: Mechanisms,
pathophysiological roles, and analysis. Biol Chem. 393:547–564.
2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Sas K, Szabó E and Vécsei L: Mitochondria,
oxidative stress and the kynurenine system, with a focus on ageing
and neuroprotection. Molecules. 23(191)2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Jagannathan R, Thapa D, Nichols CE,
Shepherd DL, Stricker JC, Croston TL, Baseler WA, Lewis SE,
Martinez I and Hollander JM: Translational regulation of the
mitochondrial genome following redistribution of mitochondrial
MicroRNA in the diabetic heart. Circ Cardiovasc Genet. 8:785–802.
2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kaliszewska A, Allison J, Martini M and
Arias N: Improving Age-related cognitive decline through dietary
interventions targeting mitochondrial dysfunction. Int J Mol Sci.
22(3574)2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Moehle EA, Shen K and Dillin A:
Mitochondrial proteostasis in the context of cellular and
organismal health and aging. J Biol Chem. 294:5396–5407.
2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Xu ZP, Yang SL, Zhao S, Zheng CH, Li HH,
Zhang Y, Huang RX, Li MZ, Gao Y, Zhang SJ, et al: Biomarkers for
early diagnostic of mild cognitive impairment in type-2 diabetes
patients: A multicentre, retrospective, nested Case-control study.
EBioMedicine. 5:105–113. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Trumpff C, Anna LM, Carla BA, James LM,
Judith EC, Gabriel S, Amy EV, Eugene VM, Zhenglong G, Brett AK and
Martin P: Acute psychological stress increases serum circulating
cell-free mitochondrial DNA. Psychoneuroendocrinology. 106:268–276.
2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Labbadia J, Brielmann RM, Neto MF, Lin YF,
Haynes CM and Morimoto RI: Mitochondrial stress restores the heat
shock response and prevents proteostasis collapse during aging.
Cell Rep. 21:1481–1494. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Silzer T, Robert B, Jie S, Gita P, Leigh
J, Sid O'B and Nicole P: Circulating Mitochondrial DNA: New indices
of type 2 diabetes-related cognitive impairment in Mexican
Americans. PLoS One. 14(e0213527)2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Horejs CM: Lysosomes uncovered. Nat Rev
Mater. 4(2)2019.
|
|
60
|
Holstege H, van der Lee SJ, Hulsman M,
Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin
N, Uitterlinden AG, et al: Characterization of pathogenic SORL1
genetic variants for association with Alzheimer's disease: A
clinical interpretation strategy. Eur J Hum Genet. 25:973–981.
2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Forlenza OV, Diniz BS, Teixeira AL, Stella
F and Gattaz W: Mild cognitive impairment (part 2): Biological
markers for diagnosis and prediction of dementia in Alzheimer's
disease. Rev Bras Psiquiatr. 35:284–294. 2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Klaips CL, Jayaraj GG and Hartl FU:
Pathways of cellular proteostasis in aging and disease. J Cell
Biol. 217:51–63. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Giau VV, Bagyinszky E and An SSA:
Potential fluid biomarkers for the diagnosis of mild cognitive
impairment. Int J Mol Sci. 20(4149)2019.PubMed/NCBI View Article : Google Scholar
|