1. Introduction
Thalassemia, an inherited disease, is characterized
by deficient or absent α- or β-globin chains within red blood
cells, leading to imbalanced globin chains and reduced red cell
survival. Excess globin chains form hemichromes, causing oxidative
stress and phosphatidylserine (PS) exposure on red cell membranes
(1,2). Thalassemia varies in severity:
Thalassemia minor, thalassemia intermedia (TI) and thalassemia
major (TM). Patients with TM require regular blood transfusions
commencing within the first 2 years of life. Untreated or
inadequately transfused patients display jaundice, growth
retardation, splenohepatomegaly and skeletal malformation from
extramedullary hematopoiesis. Excessive iron from transfusions may
lead to irreversible organ damage, with death often resulting from
cardiovascular diseases, iron overload and infections (3-5).
Of note, patients with β-thalassemia have an
increased risk of hypercoagulability and thrombosis, significantly
impacting morbidity and mortality (6). This phenomenon involves interactions
between damaged red cells, activated leukocytes and platelets,
adhesive endothelial cells and coagulation factor dysregulation
(7). The incidence of thrombosis
may be minimized by regular blood transfusion, while splenectomy
significantly potentiates the risk (8,9).
Increased circulating cell-derived vesicles have been reported in
splenectomized thalassemic patients, suggesting the role of the
spleen in the clearance of these vesicles (10).
Extracellular vesicles (EVs) are submicron-sized,
bioactive membrane vesicles released by cells under stress,
activation or apoptosis. They are classified into three types:
Apoptotic bodies, microparticles (MPs), which are large EVs, and
exosomes (which are small EVs), each differing in their biogenesis,
properties and molecular markers. They have received extensive
interest due to their important role as a conveyer of biological
cargos. EVs are critical in cell communication, carrying proteins,
lipids and nucleic acids to target cells (11,12).
Their elevated levels in cardiovascular and hematological disorders
make them potential diagnostic and prognostic biomarkers (12-14).
Our group in Thailand was the first to characterize MPs in patients
with β-thalassemia/hemoglobin (Hb)E following bone marrow
transplantation. Lower levels of circulating PS-externalizing red
blood cells, their MPs and procoagulant platelets were found in
transplanted patients than in those receiving regular transfusions
(15). These circulating MPs, along
with leukocyte-platelet aggregates, may contribute to
hypercoagulability in patients with β-thalassemia/HbE who have
undergone bone marrow transplantation (16). Understanding the role of EVs in
altering target cell phenotypes and inducing functional changes may
provide insight into the disease's pathogenesis and
complications.
2. β-Thalassemia
β-Thalassemia, an autosomal recessive anemia, arises
from reduced (β+) or absent (β0) synthesis of
the β-globin chain. Its severity varies, primarily depending on the
excessive degree of α-globin chains, which accumulate in red blood
cell precursors, causing ineffective erythropoiesis (17). Ineffective erythropoiesis features
rapid expansion of early erythroid precursors and apoptosis of
late-stage ones, resulting in low production of reticulocytes and
mature red blood cells. This mechanism is primarily responsible for
inherited anemia disorders such as β-thalassemia (18).
β-Thalassemia is categorized into β-TM, β-TI,
thalassemia minor and the β-thalassemia trait. Patients with TM
require regular blood transfusions for survival. Untreated or
poorly transfused individuals exhibit growth retardation, pallor,
jaundice, skin pigmentation, weak musculature, hepatosplenomegaly,
leg ulcers, extramedullary hematopoiesis masses and skeletal
changes due to marrow expansion.
Patients who do not receive blood transfusions
usually succumb to heart failure. However, those receiving
transfusions may develop iron overload, depending on their
adherence to iron chelation therapy. Complications related to iron
overload in children include growth retardation and failure of
sexual maturation. In adults, complications may include liver
fibrosis and cirrhosis, endocrine gland involvement (e.g., diabetes
mellitus, and insufficiency of hormones released from the
parathyroid, thyroid and pituitary glands), and cardiovascular
diseases such as dilated myocardiopathy and arrhythmias (19).
Patients with TI have milder anemia, requiring
infrequent or no transfusions. They may be asymptomatic until
adulthood, with symptoms including pallor, mild jaundice,
cholelithiasis, liver and spleen enlargement, bone changes, leg
ulcers, extramedullary erythroid marrow masses, osteopenia,
osteoporosis and thrombotic complications (17). Patients with TI often have an
increased tendency to develop thrombosis, particularly when
compared to those with TM, particularly in splenectomized patients.
This increased risk may lead to thromboembolic conditions such as
deep vein thrombosis, portal vein thrombosis, stroke and pulmonary
embolism (20). The β-thalassemia
trait is typically clinically asymptomatic, although mild anemia
may occur in certain cases (17).
The high incidence of thromboembolic events in TI
leads to a chronic hypercoagulable state, causing major morbidity
and mortality. These events include cerebral thrombosis, deep
venous thrombosis, pulmonary embolism and recurrent arterial
occlusion (7). In addition, all
studies of EVs in β-thalassemia classified the patients as TM, and
TI according to severity, anemia, blood transfusion and
complications due to the higher incidence of hypercoagulable state
found in patients with TI. In the present review, the forms of
β-thalassemia, such as TM, TI and β-thalassemia trait, were
described.
3. α-Thalassemia
In α-thalassemia, the clinical phenotypes range from
asymptomatic to lethal, depending on the number of non-functional
copies of α-globin genes. Two main groups of α-thalassemia include
α-thalassemia 1 and α-thalassemia 2. As a result of deficient
production of α-globin, it leads to the formation of homotetramer
of unaffected β-globin chains. One of the moderately severe
symptomatic forms of α-thalassemia is HbH disease (β4
tetramers). The non-deletion (ND) form of HbH disease
(ααND/-) and Hb constant spring, which is
caused by mutation in α-globin gene termination, are common in
Asian countries. Patients in this group are severely anemic and
have a risk of iron overload and splenomegaly. Therefore,
splenectomy is recommended for patients with ND HbH, but they have
a high rate of counteracting with the pro-thrombophilic
complication (2,21). Similarly, abnormal externalization
of PS on the red cell surface and their shedding MPs can stimulate
the disturbance of coagulation. However, the coagulation parameters
tested by Sirachainan et al (22) were not different among pediatric
patients with α-thalassemia and normal controls. Further
investigations on the effect of other factors, such as activation
of platelet and endothelial cells and shedding EV, are required to
confirm their contribution of hemostatic alteration in
α-thalassemia.
4. EVs in patients with thalassemia
In patients with thalassemia, various studies have
examined the profile and quantity of circulating MPs, focusing on
their cellular origin and procoagulant properties. Among these
studies, those involving patients who have undergone splenectomy
revealed a notable presence of plasma MPs, primarily released from
platelets and damaged red cells, which express negatively charged
PS (23). In patients with
β-thalassemia/HbE who have been splenectomized, these MPs are known
to trigger platelet activation and aggregation, contributing to the
formation of thrombi (24). The
notably elevated levels of these MPs may partly contribute to
thromboembolic events, hypercoagulable states and other clinical
complications commonly observed in patients with thalassemia
(25).
Discovery of EVs in thalassemia
The etiology of thromboembolic risk in thalassemic
patients involves several factors. They include hyperaggregation
and oxidative damage of red cells, chronic platelet activation and
increased circulating cell-derived EVs. This complex interplay
requires further investigation to fully understand its underlying
mechanisms. The initial functional exploration of the role of EVs
in the pathogenesis of the hypercoagulable state in thalassemia
involved coculturing recipient cells, such as endothelial cells and
leukocytes, with isolated EVs. This was followed by analyzing the
induced phenotypic changes in these target cells to gain insight
into the impact of EVs.
Initial evidence of the procoagulant role of EVs and
their influence on thromboembolic events and endothelial
dysfunction came from observing endothelial cells interacting with
PS-externalizing red cells. This exposure showed that PS-enriched
lipid vesicles compete for PS-binding sites on endothelial cell
monolayers, indicating the prothrombotic effects of both PS-exposed
red cells and PS-enriched vesicles (26). Studies have documented significant
evidence that red cells in thalassemic patients externalize PS on
their outer surface (27,28). When these cells shed as
PS-expressing red cell vesicles, they contribute to the initiation
of chronic hypercoagulability, along with other procoagulant
factors (29-31).
With advancements in flow cytometric analysis,
Pattanapanyasat et al (32)
and another study by the same group (33) successfully quantified the number of
vesicles shed from red cells. These vesicles are phenotypically
distinct, as identified by their reactivity to annexin-V coupled
with glycophorin A (CD235a). They are also smaller than the
platelet population, as observed in logarithmic forward- and
side-scatter dot plots. These characteristics revealed that
thalassemic patients have more circulating red cell vesicles than
healthy individuals. Furthermore, patients who had undergone
splenectomy were found to have significantly elevated levels of
these vesicles.
These results align with those reported by other
research groups, who found that platelets, rather than red
cell-derived EVs, are predominantly responsible for procoagulant
activity in splenectomized patients receiving blood transfusions
(34). In addition, a marked
increase in platelet-derived EVs has been identified as a
predictive biomarker for thromboembolic events in both patients
with TM and TI (35,36).
In addition to red blood cells and platelets, other
cell types also release EVs. Flow cytometry may be employed to
identify the cellular origin of these EVs using
fluorochrome-conjugated antibodies specific to their parent cells
(37). Common surface markers used
to determine the cellular origin of EVs are listed in Table I. Beyond flow cytometry, several
other techniques are utilized for characterizing circulating EVs,
including proteomic studies for specific marker identification.
Several common biomarkers for exosomes, including major
histocompatibility complex, flotillin and the heat-shock 70-kDa
protein (HSP70) are similarly present in all EV types. Proteins
particularly enriched in small EVs and showing varying expression
levels in different EV populations have been documented. Western
blot analysis is used to assess the differential expression of CD9,
CD63 and CD81. These transmembrane proteins are frequently observed
in EVs from various cell types, providing a framework for
identifying subtypes of EVs (38,39).
 | Table ISurface antigens employed in the
characterization of circulating extracellular vesicles using flow
cytometry. |
Table I
Surface antigens employed in the
characterization of circulating extracellular vesicles using flow
cytometry.
| Antigen marker | Alternative
name | Cell of origin |
|---|
|
Phosphatidylserine | - | Apoptotic cell
membrane |
| CD235a | Glycophorin A | Red cells |
| CD71 | Transferrin
receptor | Erythroid
precursor |
| CD41a | Glycoprotein
IIb | Platelets |
| CD42b | Glycoprotein
Ibα | Platelets |
| CD31 | Platelet
endothelial cell adhesion molecule | Platelets,
endothelial cells |
| CD144 | Vascular
endothelial cadherin (or cadherin-5) | Endothelial
cells |
| CD146 | Melanoma cell
adhesion molecule | Endothelial
cells |
| CD105 | Endoglin | Mesenchymal
stroma |
| CD45 | Protein tyrosine
phosphatase receptor type C | Leukocytes |
| CD11b | Integrin α-M | Activated
leukocytes |
| CD62P | P-selectin or
platelet activation-dependent granule membrane protein | Activated
platelets |
| CD142 | Coagulation factor
III or tissue factor | Leukocytes and
subendothelial cells, such as smooth muscle cells and
fibroblasts |
Transmission electron microscopy, a widely utilized
technique (40), is crucial in
assessing the quality and purity of samples containing EVs. This
technique can distinguish individual EVs from particles of similar
size that are not EVs (41).
In addition, microRNAs (miRNAs) associated with EVs
have gained attention as potential biomarkers for various human
diseases. To detect these EV-miRNAs, reverse
transcription-quantitative PCR (RT-qPCR) is commonly employed
(42). Recent advancements include
the development of a simplified and cost-effective approach,
single-step RT-qPCR, which allows for the direct detection of
EV-miRNAs without the need for RNA purification (43).
Nanoparticle tracking analysis is another critical
technology used for quantifying and determining the size of EVs. It
offers reproducibility and accuracy in measuring the particle
concentration and size distribution of EVs isolated from diverse
sources through various methods. To enhance reproducibility in
future EV research, standardization of nanoparticle tracking
analysis methods is essential (44).
EVs as predictive biomarkers in
thalassemia
Proteomic analysis of plasma vesicles from
β-thalassemia/HbE patients has revealed an accumulative increase in
proteins associated with oxidative damage in red blood cells and
platelets. Notable among these proteins are peroxiredoxin 6 and
HSP90(45). In addition, changes in
the levels of specific proteins in EVs from thalassemic patients,
such as increased α-Hb-stabilizing protein and decreased
haptoglobin, hemopexin and cathepsin S, suggest their potential as
biomarkers for hemolysis and inflammation. Mass spectrometry has
been utilized to quantify these markers in patients with
β-thalassemia/HbE (46). Ferru
et al (47) explored
hemichromes, composed of denatured α-globin within thalassemic red
cell EVs, that bind to band 3 protein and trigger phosphorylation
of p27Syk kinase. This process promotes red cell membrane
instability and subsequent EV shedding. They also identified a set
of HSP70 and peroxiredoxin 2 proteins that are enriched in these
EVs (47). Levin et al
(48) used nanoparticle tracking
analysis to show increased levels of thalassemic EVs following
splenectomy. These EVs have high concentrations of HSP70, which
contributes to ineffective erythropoiesis, hemolysis and disease
severity (48). Further studies
indicate that stored thalassemic blood units show overexpression of
caspase-3 and molecular chaperones such as HSP70 and DJ-1 in the EV
fraction. This finding suggests that oxidative stress affects the
physiology and aging of stored thalassemic red cells (49).
Increased amounts of circulating EVs have been
reported to be associated with clinical complications commonly
observed in patients with β-thalassemia. This increase has been
observed to positively correlate with the levels of
hypoxia-inducible factor α, a marker for tissue hypoxia,
particularly in pediatric patients with thalassemia (50). This relationship underscores the
connection between oxidative stress and the formation of EVs, which
in turn has a role in the thromboembolic phenomena frequently
observed in these patients. Significant increases in circulating
CD146+ endothelial EVs, endothelial progenitor cells and
von Willebrand factor have also been identified. These changes are
linked to the incidence of cardiovascular complications in young
patients with β-thalassemia (51).
However, elevated levels of circulating EVs have not been
associated with pulmonary arterial hypertension in splenectomized
thalassemic patients. This finding is attributed to the use of
antiplatelet drugs aimed at reducing platelet activation levels
(52).
Classifying β-thalassemia subtypes is crucial for
effective treatment and EVs have emerged as potential biomarkers
for screening the disease and differentiating among its subtypes.
Research conducted by Li et al (53) has shed light on the proteomics
profiles of plasma-derived EVs in patients with TM, TI and healthy
donors. They identified the top six proteins for the diagnosis of
patients with β-thalassemia: Complement C1s subcomponent (C1S),
clusterin (CLU), lactotransferrin, complement C1r subcomponent,
C4b-binding protein β chain and C4b-binding protein α chain. Their
study revealed distinct proteomic patterns in patients with TI and
TM. The top six proteins that showed potential in distinguishing
patients with TM from those with TI were serotransferrin (TF), Hb
subunit α (HBA1), immunoglobulin heavy constant γ 4 (IGHG4), HBB,
plasma kallikrein B1 and apolipoprotein M. Furthermore, they
proposed a model using five proteins (TF, C1S, IGHG4, CLU and HBA1)
to discriminate among the three groups. The findings suggest the
potential of EVs in subtyping and highlight the ability of
plasma-derived EVs to aid in diagnosing β-thalassemia patients
(53).
Placenta-derived EVs have emerged as promising tools
for the diagnosis of thalassemia. These EVs are detectable in
maternal circulation as early as 6 weeks of gestation, and their
release can be induced by conditions such as hypoxia and
hyperglycemia. Changes in the quantity and composition of EVs have
been observed in placental-related complications such as
preeclampsia and fetal growth restriction, highlighting their
potential as a diagnostic tool for identifying such complications
in asymptomatic pregnant women. Furthermore, in cases of Bart's
hydrops fetuses, placental hypoxia may prompt alterations in the
number or function of placenta-derived EVs. This characteristic may
offer a valuable means for early, noninvasive prenatal diagnosis.
However, standardized detection methods are needed to effectively
use placenta-derived EV detection in diagnosing, monitoring and
treating Bart's hydrops fetalis and similar placental disorders
(54).
Pathologic function of thalassemic
EVs
In thalassemic patients, an increased number of
plasma EVs has been linked to a hypercoagulable state; however,
their specific role in thalassemia is not well defined. Various
research groups have examined the biological effects of plasma EVs
isolated from thalassemic patients on different types of recipient
cells. EVs, primarily MPs, from splenectomized patients have shown
a significant capacity to activate platelets and cause
microaggregation of leukocyte platelets compared to vesicles
isolated from healthy individuals or non-splenectomized patients
(24).
The prothrombotic function of thalassemic EVs is
further evident in their contribution to endothelial cell
dysfunction. When endothelial cells internalize EVs derived from
splenectomized patients, there is an enhanced induction of
endothelial cell activation markers and proinflammatory mediators,
as well as an increased binding ability of endothelial cells to the
human monocytic leukemia cell line THP-1 (55,56).
In addition, thalassemic EVs, particularly exosomes containing
ferritin and hemichrome, have been found to promote the
hyperproliferation of cardiac cells, potentially leading to
cardiomyopathic complications commonly seen in thalassemic patients
(57).
Fig. 1 illustrates
the prothrombotic potential of thalassemic EVs in contributing to
hyperthrombophilic phenomena. Thalassemic EVs, which are mainly
released from activated platelets and damaged red cells, facilitate
the formation of microemboli: Complexes of platelets, red cells,
leukocytes and coagulation factors. These microaggregates adhere to
activated endothelial cells, triggering a hypercoagulable state in
thalassemic patients.
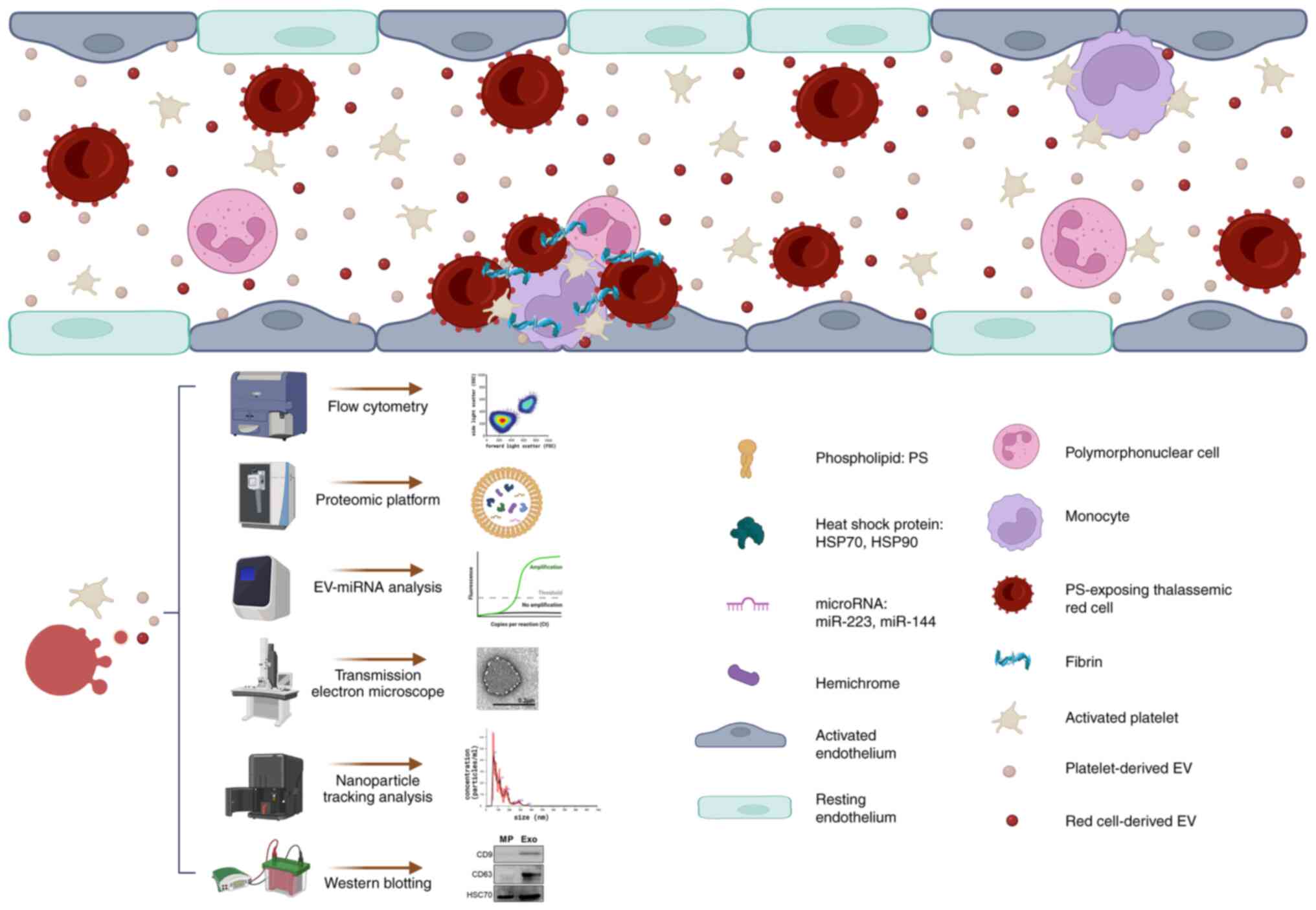 | Figure 1Role of thalassemic EVs in
thromboembolic events. The schematic illustrates how thalassemic
EVs, predominantly released by activated platelets and damaged red
cells, contribute to the formation of microemboli. These
microemboli, composed of platelets, red cells, leukocytes and
coagulation factors, adhere to activated endothelial cells, thereby
inducing a hypercoagulable state in thalassemic patients. The
characterization of these circulating EVs involves various advanced
techniques: Flow cytometry (to determine size and cell of origin);
proteomic analysis (to identify biological content); reverse
transcription-quantitative PCR (for EV miRNA profiling);
transmission electron microscopy (to assess the quality and purity
of EV-containing samples); nanoparticle tracking analysis (to
measure size and concentration); and western blot analysis (to
detect HSP70 and EV-specific protein markers, such as the
tetraspanin proteins CD9 and CD63). The figure was designed using
BioRender.com. EV, extracellular vesicle;
miRNA/miR, microRNA; HSP, heat shock protein. |
Exosomes have a critical role in intercellular
communication by shuttling biological cargos, such as miRNAs, to
recipient cells. This transfer leads to modifications in the
functional properties of target cells. In the case of
β-thalassemia, where genes that encode γ-globin are silent,
thalassemic exosomes and miR-223-3p have been reported to suppress
γ-globin expression by downregulating LIM-only protein 2, the
globin gene regulator (58).
Furthermore, differential expression of miRNA profiles in EVs from
patients with β-thalassemia compared to those from healthy
individuals has been observed.
When various recipient cells, such as endothelial
cells, hepatocytes, pancreatic cells and mesenchymal stromal cells,
are exposed to thalassemic EVs containing miR-144-3p, they exhibit
dysfunctional phenotypes. These include reduced cell viability and
induction of cell apoptosis. The mechanism behind β-thalassemia
EV-induced apoptosis in endothelial cells is associated with the
MAPK/JNK signal transduction pathway. By contrast, EVs from
splenectomized patients with β-thalassemia induce the proliferation
of bone marrow mesenchymal stromal cells. This finding suggests
that EVs contribute to organ damage and complications in
β-thalassemia (59).
EVs in bone marrow
transplantation
Allogeneic hematopoietic stem cell transplantation
(HSCT) is currently the only established, potentially curative
therapy for patients with TM. However, it is limited by the
availability of matched donors and each patient's medical
condition. Human leukocyte antigen (HLA)-matched stem cells,
sourced from cord blood or bone marrow, have yielded excellent
outcomes in HSCT. Therefore, it is recommended that patients with
TM, particularly young patients, should consider HSCT at an early
stage of the disease if they have HLA-identical sibling donors.
This approach aims to prevent the development of severe clinical
complications due to iron overload and multiorgan failure.
Anurathapan et al (60,61)
have utilized haploidentical-related donors as a source of
hematopoietic stem cells for pediatric patients with TM. Following
matched-donor or haploidentical HSCT, certain patients with
thalassemia exhibited increased endothelial activation and a
heightened risk of thromboembolic events. These complications are
attributed to several factors, including the conditioning regimen,
high-dose chemotherapy, immune response and graft-versus-host
disease (62). However, evidence
indicates that HSCT can effectively correct hemostatic alterations
in patients with TM. Sirachainan et al (63) found that plasma levels of
coagulation markers (thrombin-antithrombin complex, prothrombin
fragment and D-dimer) and anticoagulation factors (protein C,
protein S and antithrombin activity) returned to normal levels
following successful HSCT. This signified a positive therapeutic
outcome.
Several studies have noted an increase in the number
of circulating EVs following HSCT in hematological disorders.
Trummer et al (64), for
instance, reported a rise in plasma P-selectin glycoprotein
ligand-1-expressing MPs and linked this increase to the risk of
refractory disease and relapse in patients. Research by our group
focusing on EV profiling in HSCT for thalassemia, flow cytometric
analysis was utilized to measure plasma MPs in children with TM
undergoing HSCT. The randomized study by our group specifically
measured PS-expressing red cells and CD235a+ red cell
MPs. The levels of these two factors decreased after
transplantation (15). Conversely,
in another study by our group, an increase in other MP populations
from platelets (CD41a+ MP), leukocytes (CD45+
MP) and endothelial cells (CD146+ MP) in the plasma of
transplanted patients was observed. These procoagulant MPs were
associated with a higher incidence of monocyte-platelet
microaggregates, suggesting a complex interaction and potential
clinical implications in the post-transplant setting (16).
5. EVs in other hemoglobinopathies
In the realm of common hemoglobinopathies, sickle
cell disease (SCD) serves as the prototype. This inherited disorder
arises from a point mutation in the β-globin gene, leading to the
formation of hemoglobin S. SCD is often linked to chronic vascular
inflammation and hypercoagulability. These conditions result from
the combined effects of procoagulant sickle red cells, activated
platelets and cell-derived EVs. Collectively, these components
induce endothelial cell activation, leading to cardiovascular
complications (25,65,66).
Two core mechanisms have been identified in this process: The
transfer of heme to endothelial cells (67) and the increased adhesiveness of
sickle red cells to endothelial cells (68). These interactions highlight the
complex pathophysiology of SCD and the significant role of EVs in
its progression and complications.
In SCD, numerous studies have reported the presence
of large numbers of EVs, particularly those derived from red blood
cells and platelets. EVs are found in patients both during
steady-state and sickling crises, and their levels are
significantly higher than those in healthy individuals (69,70).
These sickle cell-derived EVs initiate hemostatic abnormalities,
such as prolonged thrombin generation (71). With regard to abnormal thrombin
generation in SCD, the amplification was presumably mediated by PS
exposed on sickle cell MPs that activates the intrinsic pathway of
the coagulation cascade through factor XI (69), and the extent of prothrombotic PS
relies on the size of red cell MPs, particularly macrovesicles
(72). Of note, treatments with
hydroxyurea (73) or
hydroxycarbamide (74) have been
shown to ameliorate these effects.
Beyond their role as bioeffectors in SCD, these
sickle cell-derived EVs also serve as biomarkers for predicting
disease-related complications. These complications include
oxidative stress related to vaso-occlusive crises (75), osteonecrosis (76), leg ulcers and pulmonary hypertension
(77). A notable aspect of SCD-EVs
is their content of functional miRNAs, specifically miR-124-3p,
miR-2278 and miR-4763-5p. These miRNAs are reported to be enriched
in SCD-plasma EVs and could contribute to the initiation and
progression of the disease (78).
This finding highlights the multifaceted role of EVs in the
pathophysiology and potential diagnostic and therapeutic approaches
in SCD.
6. Conclusions and future perspectives
In both normal and disease conditions, EVs,
including MPs and exosomes, are produced by most, if not all, cell
lineages. The study of MPs has garnered increased attention in
exploring the pathogenesis of common complications in patients with
thalassemia. While certain studies have demonstrated the regulatory
role of exosomes and exosomal miRNAs in contributing to
physiological and pathophysiological processes in thalassemia
biology, further research is needed. The phenotypic
characterization and functional study of EVs require precise and
accurate methodologies and techniques due to the challenges posed
by their small size. Understanding the molecular mechanisms that
govern the production and clearance of EVs, as well as those
involved in intercellular communication, is crucial. This knowledge
could pave the way for novel therapeutic approaches to reduce
clinical complications in thalassemia, offering new insight and
potential breakthroughs in the treatment of thalassemia.
Acknowledgements
Not applicable.
Funding
Funding: The authors were funded by the National Research
Council of Thailand: High-Potential Research Team Grant Program
(grant no. N42A650870), the Talented Young Researcher Scholarship
Program (grant no. N41A640122) and the Office of the Permanent
Secretary, Ministry of Higher Education, Science, Research and
Innovation (grant no. RGNS 63-161). The funders had no role in the
design of the study; the collection, analysis, or interpretation of
data; the decision to publish; or the writing of this
manuscript.
Availability of data and materials
Not applicable.
Authors' contributions
Conceptualization: PK, KP and PP. Writing-original
draft preparation: PK and PP. Writing-review and editing: KP and
PP. Figure preparation and visualization: PK. Supervision: KP and
PP. Funding acquisition: KP, PK and PP. All authors have read and
agreed to the published version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rund D and Rachmilewitz E:
Beta-thalassemia. N Engl J Med. 353:1135–1146. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Piel FB and Weatherall DJ: The
α-thalassemias. N Engl J Med. 371:1908–1916. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Borgna-Pignatti C, Rugolotto S, De Stefano
P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL,
Gamberini MR, Ghilardi R, et al: Survival and complications in
patients with thalassemia major treated with transfusion and
deferoxamine. Haematologica. 89:1187–1193. 2004.PubMed/NCBI
|
|
4
|
Zurlo MG, De Stefano P, Borgna-Pignatti C,
Di Palma A, Piga A, Melevendi C, Di Gregorio F, Burattini MG and
Terzoli S: Survival and causes of death in thalassaemia major.
Lancet. 2:27–30. 1989.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sleiman J, Tarhini A, Bou-Fakhredin R,
Saliba AN, Cappellini MD and Taher AT: Non-Transfusion-Dependent
Thalassemia: An update on complications and management. Int J Mol
Sci. 19(182)2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cappellini MD, Robbiolo L, Bottasso BM,
Coppola R, Fiorelli G and Mannucci AP: Venous thromboembolism and
hypercoagulability in splenectomized patients with thalassaemia
intermedia. Br J Haematol. 111:467–473. 2000.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Eldor A and Rachmilewitz EA: The
hypercoagulable state in thalassemia. Blood. 99:36–43.
2002.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Atichartakarn V, Angchaisuksiri P,
Aryurachai K, Chuncharunee S and Thakkinstian A: In vivo platelet
activation and hyperaggregation in hemoglobin E/beta-thalassemia: A
consequence of splenectomy. Int J Hematol. 77:299–303.
2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Atichartakarn V, Chuncharunee S,
Chandanamattha P, Likittanasombat K and Aryurachai K: Correction of
hypercoagulability and amelioration of pulmonary arterial
hypertension by chronic blood transfusion in an asplenic hemoglobin
E/beta-thalassemia patient. Blood. 103:2844–2846. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Atichartakarn V, Angchaisuksiri P,
Aryurachai K, Onpun S, Chuncharunee S, Thakkinstian A and
Atamasirikul K: Relationship between hypercoagulable state and
erythrocyte phosphatidylserine exposure in splenectomized
haemoglobin E/beta-thalassaemic patients. Br J Haematol.
118:893–898. 2002.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yáñez-Mó M, Siljander PR, Andreu Z, Zavec
AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J,
et al: Biological properties of extracellular vesicles and their
physiological functions. J Extracell Vesicles.
4(27066)2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Loyer X, Vion AC, Tedgui A and Boulanger
CM: Microvesicles as cell-cell messengers in cardiovascular
diseases. Circ Res. 114:345–353. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Westerman M, Pizzey A, Hirschman J, Cerino
M, Weil-Weiner Y, Ramotar P, Eze A, Lawrie A, Purdy G, Mackie I and
Porter J: Microvesicles in haemoglobinopathies offer insights into
mechanisms of hypercoagulability, haemolysis and the effects of
therapy. Br J Haematol. 142:126–135. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aharon A, Rebibo-Sabbah A, Tzoran I and
Levin C: Extracellular vesicles in hematological disorders. Rambam
Maimonides Med J. 5(e0032)2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Klaihmon P, Vimonpatranon S, Noulsri E,
Lertthammakiat S, Anurathapan U, Sirachainan N, Hongeng S and
Pattanapanyasat K: Normalized levels of red blood cells expressing
phosphatidylserine, their microparticles, and activated platelets
in young patients with β-thalassemia following bone marrow
transplantation. Ann Hematol. 96:1741–1747. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Klaihmon P, Lertthammakiat S, Anurathapan
U, Pakakasama S, Sirachainan N, Hongeng S and Pattanapanyasat K:
Activated platelets and leukocyte activations in young patients
with β-thalassemia/HbE following bone marrow transplantation.
Thromb Res. 169:8–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Origa R: β-Thalassemia. Genet Med.
19:609–619. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cazzola M: Ineffective erythropoiesis and
its treatment. Blood. 139:2460–2470. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Galanello R and Origa R: Beta-thalassemia.
Orphanet J Rare Dis. 5(11)2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Taher AT, Otrock ZK, Uthman I and
Cappellini MD: Thalassemia and hypercoagulability. Blood Rev.
22:283–292. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Vichinsky EP: Clinical manifestations of
alpha-thalassemia. Cold Spring Harb Perspect Med.
3(a011742)2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sirachainan N, Chuansumrit A, Kadegasem P,
Sasanakul W, Wongwerawattanakoon P and Mahaklan L: Normal
hemostatic parameters in children and young adults with
α-thalassemia diseases. Thromb Res. 146:35–42. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pattanapanyasat K, Gonwong S, Chaichompoo
P, Noulsri E, Lerdwana S, Sukapirom K, Siritanaratkul N and
Fucharoen S: Activated platelet-derived microparticles in
thalassaemia. Br J Haematol. 136:462–471. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Klaihmon P, Phongpao K, Kheansaard W,
Noulsri E, Khuhapinant A, Fucharoen S, Morales NP, Svasti S,
Pattanapanyasat K and Chaichompoo P: Microparticles from
splenectomized β-thalassemia/HbE patients play roles on
procoagulant activities with thrombotic potential. Ann Hematol.
96:189–198. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tantawy AA, Adly AA, Ismail EA, Habeeb NM
and Farouk A: Circulating platelet and erythrocyte microparticles
in young children and adolescents with sickle cell disease:
Relation to cardiovascular complications. Platelets. 24:605–614.
2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Manodori AB, Barabino GA, Lubin BH and
Kuypers FA: Adherence of phosphatidylserine-exposing erythrocytes
to endothelial matrix thrombospondin. Blood. 95:1293–1300.
2000.PubMed/NCBI
|
|
27
|
Zahedpanah M, Azarkeivan A, Aghaieepour M,
Nikogoftar M, Ahmadinegad M, Hajibeigi B, Tabatabaiee MR and
Maghsudlu M: Erythrocytic phosphatidylserine exposure and
hemostatic alterations in beta-thalassemia intermediate patients.
Hematology. 19:472–476. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mahdi ZN, Al-Mudallal SS and Hameed BM:
Role of red blood cells ‘annexin V’ and platelets ‘P-selectin’ in
patients with thalassemia. Hematol Oncol Stem Cell Ther. 12:15–18.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chung SM, Bae ON, Lim KM, Noh JY, Lee MY,
Jung YS and Chung JH: Lysophosphatidic acid induces thrombogenic
activity through phosphatidylserine exposure and procoagulant
microvesicle generation in human erythrocytes. Arterioscler Thromb
Vasc Biol. 27:414–421. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Willekens FL, Were JM, Groenen-Döpp YA,
Roerdinkholder-Stoelwinder B, de Pauw B and Bosman GJ: Erythrocyte
vesiculation: A self-protective mechanism? Br J Haematol.
141:549–556. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Camus SM, Gausserès B, Bonnin P, Loufrani
L, Grimaud L, Charue D, De Moraes JA, Renard JM, Tedgui A,
Boulanger CM, et al: Erythrocyte microparticles can induce kidney
vaso-occlusions in a murine model of sickle cell disease. Blood.
120:5050–5058. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pattanapanyasat K, Noulsri E, Fucharoen S,
Lerdwana S, Lamchiagdhase P, Siritanaratkul N and Webster HK: Flow
cytometric quantitation of red blood cell vesicles in thalassemia.
Cytometry B Clin Cytom. 57:23–31. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lamchiagdhase P, Nitipongwanich R,
Rattanapong C, Noulsri E, Lerdwana S and Pattanapanyasat K: Red
blood cell vesicles in thalassemia. J Med Assoc Thai. 87:233–238.
2004.PubMed/NCBI
|
|
34
|
Agouti I, Cointe S, Robert S, Judicone C,
Loundou A, Driss F, Brisson A, Steschenko D, Rose C, Pondarré C, et
al: Platelet and not erythrocyte microparticles are procoagulant in
transfused thalassaemia major patients. Br J Haematol. 171:615–624.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Youssry I, Soliman N, Ghamrawy M, Samy RM,
Nasr A, Abdel Mohsen M, ElShahaat M, Bou Fakhredin R and Taher A:
Circulating microparticles and the risk of thromboembolic events in
Egyptian beta thalassemia patients. Ann Hematol. 96:597–603.
2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Habib A, Kunzelmann C, Shamseddeen W,
Zobairi F, Freyssinet JM and Taher A: Elevated levels of
circulating procoagulant microparticles in patients with
beta-thalassemia intermedia. Haematologica. 93:941–942.
2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nielsen MH, Beck-Nielsen H, Andersen MN
and Handberg A: A flow cytometric method for characterization of
circulating cell-derived microparticles in plasma. J Extracell
Vesicles. 3:2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kowal J, Arras G, Colombo M, Jouve M,
Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M and Théry
C: Proteomic comparison defines novel markers to characterize
heterogeneous populations of extracellular vesicle subtypes. Proc
Natl Acad Sci USA. 113:E968–E977. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kowal EJK, Ter-Ovanesyan D, Regev A and
Church GM: Extracellular Vesicle Isolation and Analysis by Western
Blotting. Methods Mol Biol. 1660:143–152. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tijssen MR, Cvejic A, Joshi A, Hannah RL,
Ferreira R, Forrai A, Bellissimo DC, Oram SH, Smethurst PA, Wilson
NK, et al: Genome-wide analysis of simultaneous GATA1/2, RUNX1,
FLI1, and SCL binding in megakaryocytes identifies hematopoietic
regulators. Dev Cell. 20:597–609. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rikkert LG, Nieuwland R, Terstappen Lwmm
and Coumans FAW: Quality of extracellular vesicle images by
transmission electron microscopy is operator and protocol
dependent. J Extracell Vesicles. 8(1555419)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
El Andaloussi S, Mäger I, Breakefield XO
and Wood MJA: Extracellular vesicles: Biology and emerging
therapeutic opportunities. Nat Rev Drug Discov. 12:347–357.
2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lee H, He X, Le T, Carnino JM and Jin Y:
Single-step RT-qPCR for detection of extracellular vesicle
microRNAs in vivo: A time- and cost-effective method. Am J Physiol
Lung Cell Mol Physiol. 318:L742–l749. 2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Comfort N, Cai K, Bloomquist TR, Strait
MD, Ferrante AW Jr and Baccarelli AA: Nanoparticle tracking
analysis for the quantification and size determination of
extracellular vesicles. J Vis Exp: Mar 28, 2021 (Epub ahead of
print). doi: 10.3791/62447.
|
|
45
|
Chaichompoo P, Kumya P, Khowawisetsut L,
Chiangjong W, Chaiyarit S, Pongsakul N, Sirithanaratanakul N,
Fucharoen S, Thongboonkerd V and Pattanapanyasat K:
Characterizations and proteome analysis of platelet-free
plasma-derived microparticles in β-thalassemia/hemoglobin E
patients. J Proteomics 76 Spec No.: 239-250, 2012.
|
|
46
|
Kittivorapart J, Crew VK, Wilson MC,
Heesom KJ, Siritanaratkul N and Toye AM: Quantitative proteomics of
plasma vesicles identify novel biomarkers for hemoglobin
E/β-thalassemic patients. Blood Adv. 2:95–104. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ferru E, Pantaleo A, Carta F, Mannu F,
Khadjavi A, Gallo V, Ronzoni L, Graziadei G, Cappellini MD and
Turrini F: Thalassemic erythrocytes release microparticles loaded
with hemichromes by redox activation of p72Syk kinase.
Haematologica. 99:570–578. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Levin C, Koren A, Rebibo-Sabbah A, Koifman
N, Brenner B and Aharon A: Extracellular Vesicle Characteristics in
β-thalassemia as Potential Biomarkers for Spleen Functional Status
and Ineffective Erythropoiesis. Front Physiol.
9(1214)2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tzounakas VL, Anastasiadi AT,
Dzieciatkowska M, Karadimas DG, Stamoulis K, Papassideri IS, Hansen
KC, D'Alessandro A, Kriebardis AG and Antonelou MH: Proteome of
Stored RBC membrane and vesicles from heterozygous beta thalassemia
donors. Int J Mol Sci. 22(3369)2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Elsayh KI, Zahran AM, El-Abaseri TB,
Mohamed AO and El-Metwally TH: Hypoxia biomarkers, oxidative
stress, and circulating microparticles in pediatric patients with
thalassemia in Upper Egypt. Clin Appl Thromb Hemost. 20:536–545.
2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Adly AA, El-Sherif NH, Ismail EA, El-Zaher
YA, Farouk A, El-Refaey AM and Wahba MS: Vascular dysfunction in
patients with young β-thalassemia: Relation to cardiovascular
complications and subclinical atherosclerosis. Clin Appl Thromb
Hemost. 21:733–744. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Manakeng K, Prasertphol P, Phongpao K,
Chuncharunee S, Tanyong D, Worawichawong S, Svasti S and
Chaichompoo P: Elevated levels of platelet- and red cell-derived
extracellular vesicles in transfusion-dependent β-thalassemia/HbE
patients with pulmonary arterial hypertension. Ann Hematol.
98:281–288. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Li N, Wu B, Wang J, Yan Y, An P, Li Y, Liu
Y, Hou Y, Qing X, Niu L, et al: Differential proteomic patterns of
plasma extracellular vesicles show potential to discriminate
β-thalassemia subtypes. iScience. 26(106048)2023.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Chaemsaithong P, Luewan S, Taweevisit M,
Chiangjong W, Pongchaikul P, Thorner PS, Tongsong T and
Chutipongtanate S: Placenta-Derived extracellular vesicles in
pregnancy complications and prospects on a liquid biopsy for
hemoglobin Bart's Disease. Int J Mol Sci. 24(5658)2023.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kheansaard W, Phongpao K, Paiboonsukwong
K, Pattanapanyasat K, Chaichompoo P and Svasti S: Microparticles
from β-thalassaemia/HbE patients induce endothelial cell
dysfunction. Sci Rep. 8(13033)2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Klaihmon P, Khuhapinant A, Kheansaard W
and Pattanapanyasat K: Internalization of cell-derived
microparticles triggers endothelial pro-inflammatory responses.
Asian Pac J Allergy Immunol: Apr 18, 2021 (Epub ahead of
print).
|
|
57
|
Atipimonpat A, Siwaponanan P, Khuhapinant
A, Svasti S, Sukapirom K, Khowawisetsut L and Pattanapanyasat K:
Extracellular vesicles from thalassemia patients carry
iron-containing ferritin and hemichrome that promote cardiac cell
proliferation. Ann Hematol. 100:1929–1946. 2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Sun KT, Huang YN, Palanisamy K, Chang SS,
Wang IK, Wu KH, Chen P, Peng CT and Li CY: Reciprocal regulation of
ү-globin expression by exo-miRNAs: Relevance to ү-globin silencing
in β-thalassemia major. Sci Rep. 7(202)2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Levin C, Koren A, Rebibo-Sabbah A, Levin
M, Koifman N, Brenner B and Aharon A: Extracellular Vesicle
MicroRNA That Are Involved in β-Thalassemia complications. Int J
Mol Sci. 22(9760)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Anurathapan U, Hongeng S, Pakakasama S,
Sirachainan N, Songdej D, Chuansumrit A, Charoenkwan P,
Jetsrisuparb A, Sanpakit K, Rujkijyanont P, et al: Hematopoietic
stem cell transplantation for homozygous β-thalassemia and
β-thalassemia/hemoglobin E patients from haploidentical donors.
Bone Marrow Transplant. 51:813–818. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Anurathapan U, Hongeng S, Pakakasama S,
Songdej D, Sirachainan N, Pongphitcha P, Chuansumrit A, Charoenkwan
P, Jetsrisuparb A, Sanpakit K, et al: Hematopoietic stem cell
transplantation for severe thalassemia patients from haploidentical
donors using a novel conditioning regimen. Biol Blood Marrow
Transplant. 26:1106–1112. 2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Lertthammakiat S, Sitthirat P, Anurathapan
U, Songdej D, Pakakasama S, Chuansumrit A, Putawornsub N,
Sirasittikarn S, Wantanawijarn S, Kadegasem P, et al: No
differences in hemostatic and endothelial activations between
haploidentical and matched-donor hematopoietic stem cell
transplantation in thalassemia disease. Thromb J.
18(21)2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Sirachainan N, Thongsad J, Pakakasama S,
Hongeng S, Chuansumrit A, Kadegasem P, Tirakanjana A, Archararit N
and Sirireung S: Normalized coagulation markers and anticoagulation
proteins in children with severe β-thalassemia disease after stem
cell transplantation. Thromb Res. 129:765–770. 2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Trummer A, De Rop C, Stadler M, Ganser A
and Buchholz S: P-selectin glycoprotein ligand-1 positive
microparticles in allogeneic stem cell transplantation of
hematologic malignancies. Exp Hematol. 9:1047–1055. 2011.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Ataga KI, Cappellini MD and Rachmilewitz
EA: Beta-thalassaemia and sickle cell anaemia as paradigms of
hypercoagulability. Br J Haematol. 139:3–13. 2007.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Garnier Y, Ferdinand S, Garnier M, Cita
KC, Hierso R, Claes A, Connes P, Hardy-Dessources MD, Lapouméroulie
C, Lemonne N, et al: Plasma microparticles of sickle patients
during crisis or taking hydroxyurea modify endothelium inflammatory
properties. Blood. 136:247–256. 2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Camus SM, De Moraes JA, Bonnin P, Abbyad
P, Le Jeune S, Lionnet F, Loufrani L, Grimaud L, Lambry JC, Charue
D, et al: Circulating cell membrane microparticles transfer heme to
endothelial cells and trigger vasoocclusions in sickle cell
disease. Blood. 125:3805–3814. 2015.PubMed/NCBI View Article : Google Scholar
|
|
68
|
An R, Man Y, Cheng K, Zhang T, Chen C,
Wang F, Abdulla F, Kucukal E, Wulftange WJ, Goreke U, et al: Sickle
red blood cell-derived extracellular vesicles activate endothelial
cells and enhance sickle red cell adhesion mediated by von
Willebrand factor. Br J Haematol. 201:552–563. 2023.PubMed/NCBI View Article : Google Scholar
|
|
69
|
van Beers EJ, Schaap MC, Berckmans RJ,
Nieuwland R, Sturk A, van Doormaal FF, Meijers JC and Biemond BJ:
CURAMA study group. Circulating erythrocyte-derived microparticles
are associated with coagulation activation in sickle cell disease.
Haematologica. 94:1513–1519. 2009.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Nebor D, Bowers A, Connes P,
Hardy-Dessources MD, Knight-Madden J, Cumming V, Reid M and Romana
M: Plasma concentration of platelet-derived microparticles is
related to painful vaso-occlusive phenotype severity in sickle cell
anemia. PLoS One. 9(e87243)2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Gerotziafas GT, Van Dreden P, Chaari M,
Galea V, Khaterchi A, Lionnet F, Stankovic-Stojanovic K,
Blanc-Brude O, Woodhams B, Maier-Redelsperger M, et al: The
acceleration of the propagation phase of thrombin generation in
patients with steady-state sickle cell disease is associated with
circulating erythrocyte-derived microparticles. Thromb Haemost.
107:1044–1052. 2012.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Smith RA, Mankelow TJ, Drizou D, Bullock
T, Latham T, Trompeter S, Blair A and Anstee DJ: Large red
cell-derived membrane particles are major contributors to
hypercoagulability in sickle cell disease. Sci Rep.
11(11035)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Nouraie M, Lee JS, Zhang Y, Kanias T, Zhao
X, Xiong Z, Oriss TB, Zeng Q, Kato GJ, Gibbs JS, et al: The
relationship between the severity of hemolysis, clinical
manifestations and risk of death in 415 patients with sickle cell
anemia in the US and Europe. Haematologica. 98:464–472.
2013.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Garnier Y, Ferdinand S, Connes P, Garnier
M, Etienne-Julan M, Lemonne N and Romana M: Decrease of
externalized phosphatidylserine density on red blood cell-derived
microparticles in SCA patients treated with hydroxycarbamide. Br J
Haematol. 182:448–451. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Hierso R, Lemonne N, Villaescusa R,
Lalanne-Mistrih ML, Charlot K, Etienne-Julan M, Tressières B,
Lamarre Y, Tarer V, Garnier Y, et al: Exacerbation of oxidative
stress during sickle vaso-occlusive crisis is associated with
decreased anti-band 3 autoantibodies rate and increased red blood
cell-derived microparticle level: A prospective study. Br J
Haematol. 176:805–813. 2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Marsh A, Schiffelers R, Kuypers F, Larkin
S, Gildengorin G, van Solinge W and Hoppe C: Microparticles as
biomarkers of osteonecrosis of the hip in sickle cell disease. Br J
Haematol. 168:135–138. 2015.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Olatunya OS, Lanaro C, Longhini AL,
Penteado CFF, Fertrin KY, Adekile A, Saad STO and Costa FF: Red
blood cells microparticles are associated with hemolysis markers
and may contribute to clinical events among sickle cell disease
patients. Ann Hematol. 98:2507–2521. 2019.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Khalyfa A, Khalyfa AA, Akbarpour M, Connes
P, Romana M, Lapping-Carr G, Zhang C, Andrade J and Gozal D:
Extracellular microvesicle microRNAs in children with sickle cell
anaemia with divergent clinical phenotypes. Br J Haematol.
174:786–798. 2016.PubMed/NCBI View Article : Google Scholar
|














