1. Introduction
Glycosylphosphatidylinositol (GPI) serves as a
glycolipid anchor for numerous cell surface proteins in eukaryotes
(1). In humans, ~150 GPI-anchored
proteins (APs) have been identified, displaying roles such as
enzymatic activity, antigen presentation, co-receptor engagement,
adhesion and involvement in immune responses (1-3).
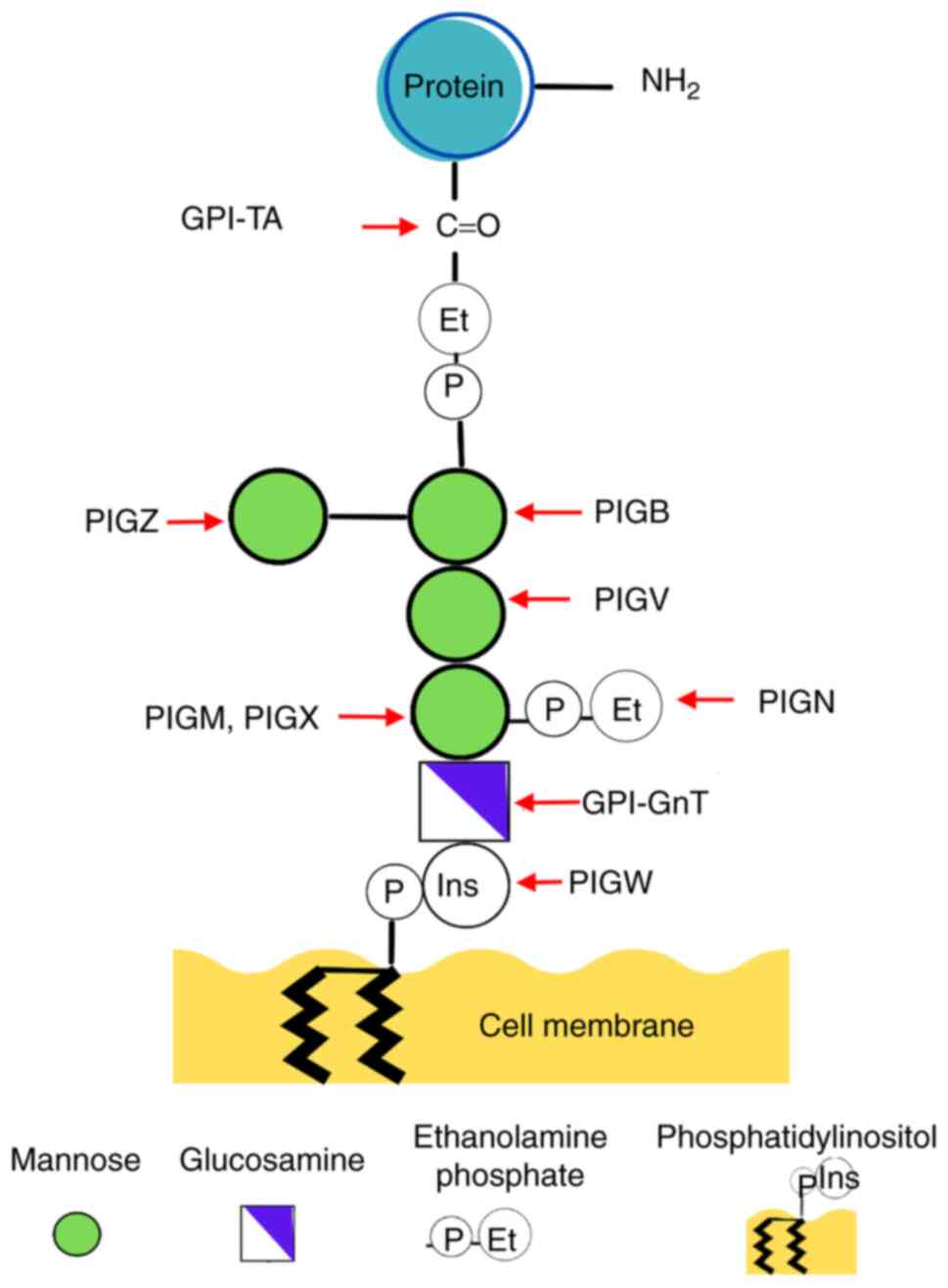
The core structure of GPI remains highly conserved
across diverse organisms, featuring a PI molecule firmly integrated
into the cell membrane. This structure is followed by glycans,
namely glucosamine (GlcN) and three mannoses (Man) culminating in a
terminal ethanolamine phosphate (EtNP) group. The latter binds
covalently to the C-terminal end of the target protein (1,4).
Variations in GPI backbone, involving modifications in EtNP and
various glycan side branches, depend on the organism, cell type and
specific protein (1,4). Notably, in mammalian cells, Man1
undergoes modification with an EtNP side chain and Man4 may attach
to Man3 via α1,2-linkage (4,5).
To date, 30 genes have been identified encoding
proteins implicated in GPI synthesis (4,6). These
genes belong to the phosphatidylinositol-glycan biosynthesis class
(PIG) and the post-GPI attachment to proteins (PGAP)
families (3). The initiation of
glycolipid biosynthesis occurs on the cytoplasmic side of the
endoplasmic reticulum (ER). GPI N-acetylglucosamine transferase
(GPI-GnT) catalyzes transfer of N-acetylglucosamine (GlcNAc) glycan
to the PI molecule within the ER membrane, yielding GlcNAc-PI
(1). Subsequent steps involve
deacetylation, translocation to the ER lumen and acylation of
inositol resulting in GlcN-acyl-PI and sequential addition of three
Man. The addition of Man1 in ER is orchestrated by
GPI-mannosyltransferase (MT)-I, anchored in the ER membrane and
composed of the proteins PIGM and PIGX (Fig. 1) (1), where PIGM serves a functional role and
PIGX as the stabilizing protein for PIGM enzyme (1).
Following GPI synthesis, GPI-MT-II encoded by
PIGV links Man2 to Man-GlcN-acyl-PI, while GPI-MT-III
encoded by PIGB adds a third Man yielding the glycolipid
Man-Man-Man3-GlcN-acyl-PI (Fig. 1)
(1,7). In addition, an EtNP molecule is
integrated into the glycan, contributing to formation of the GPI
core (1,7). The addition of Man4 as a side chain to
Man3 is facilitated by GPI-MT-IV, encoded by PIGZ (1,7).
Subsequently, the GPI molecule can bind to proteins featuring a
C-terminal GPI attachment hydrophobic signal peptide, mediated by
the GPI transamidase enzyme complex (GPI-TA) bound to the EtNP
molecule (1). Synthesis proceeds in
the Golgi apparatus, where GPI undergoes further lipid remodeling
and glycan modification by PGAP enzymes (1,7-9).
For example, the enzyme GPI-N-acetylgalactosamine transferase
(GalNAc), encoded by the PGAP4 gene, modifies Man1 by adding
a GalNAc (10). Ultimately, GPI-APs
are transported to the plasma membrane via vesicular transport
where they function within lipid rafts or are released into the
extracellular space (8). GPI-APs
exhibit diverse roles, including enzymatic activity, signaling,
cell adhesion, cell wall metabolism, neuritogenesis and immune
response (11).
Alterations in genes involved in GPI biosynthesis
have been implicated in congenital anomalies such as multiple
congenital anomalies-hypotonia-seizures, hyperphosphatasia with
mental retardation and anomalies/epilepsy syndrome (3,12,13).
In addition, certain types of cancer display altered expression in
some PIG genes (14-16).
Currently, little is known about the involvement of altered
expression of PIGM and PIGX, genes that encode and
regulate the GPI-MT-I, respectively, in human disease (17). The characteristics of the
PIGM and PIGX genes and their encoded proteins are
summarized in the present review, as well as the relevance of both
genes in GPI synthesis and certain human health diseases, and their
potential role in other biological functions.
2. Characteristics of the coding sequences
of PIGM and PIGX and their expression in humans
PIGM is localized in chromosome 1q23,
consists of 7,038 bp and encodes the transcript ENST00000368090.5,
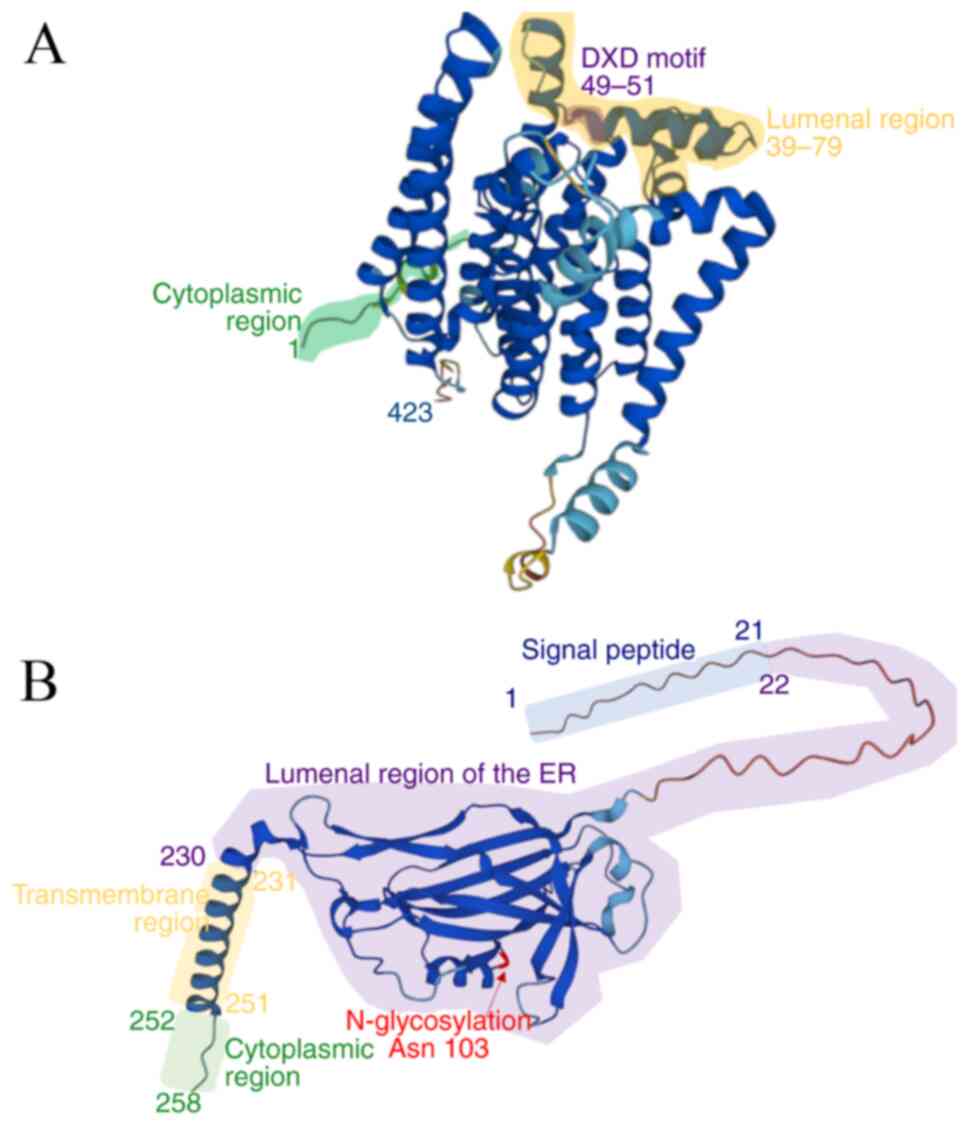
resulting in the protein PIGM Q9H3S5(18). The protein consists of 423 amino
acid residues (2); the predicted
structure by AlphaFold DB indicates that the tertiary structure
consists of 10 transmembrane α-helices that alternate with 11
lumenal and cytoplasmic domains (Fig.
2A) (19). At amino acid
positions 49-51, the protein harbors a sugar-binding motif,
aspartate-any residue-aspartate (DXD) situated within a hydrophilic
region flanked by the first and second transmembrane domains. The
DXD motif is a prevalent feature in numerous glycosyltransferases
and serves a pivotal role in coordinating a manganese ion, crucial
for binding to a nucleotide sugar substrate (20). Notably, mutations in the DXD motif,
such as the D51A alteration in PIGM, lead to the absence of GPI-APs
on the cell surface, indicating the essential role of the DXD motif
in expression of GPI-APs (20).
Moreover, according to the predicted structure, all lumenal domains
are comprised of the amino acid residues 39-79, 162-169, 247-287,
338 and 379-384, where PIGM should exert the catalytic activity
(19); to the best of our knowledge
however, there is no experimental evidence regarding the functional
importance of these regions. There is no predicted site for
phosphorylation (2) and thus far,
binding to PIGX is the only mechanism proposed to regulate
catalytic activity.
PIGX is 23,630-bp long and is located on
chromosome 3q29(21). Currently,
there is a total of nine known potential mRNA variants of
PIGX resulting from alternative splicing (18). However, only two mRNA variants
encode two protein isoforms (18).
The protein isoform of PIGX Q8TBF5-1 is encoded by mRNA variant
ENST00000392391.9, with a size of 258 amino acid residues and a
mass of 28,788 Da (2). This isoform
has been chosen as the canonical protein since it was first
described (2,18). The amino acid residues 1-21 comprise
the signal peptide that recognizes the protein as an ER membrane
protein (Fig. 2B). The amino acid
residues 22-230 form a soluble amino acid chain in the lumen of the
ER, while the amino acid residues 231-251 are inserted in the ER
membrane and amino acid residues in the C-terminal region, 252-258,
are soluble in the cytoplasm (Fig.
2B) (2). PIGX has a
non-ATG start codon and instead contains a CTG start codon that is
well-conserved in mammals (18);
non-ATG start codons are associated with key cellular functions
such as development and stress responses (22). Regarding post-translational
modifications, PIGX Q8TBF5-1 isoform harbors an N-glycosylation
site at asparagine 103, phosphorylation site at serine 136(2) and two ubiquitination sites at lysines
66 and 82(23). By contrast, the
isoform Q8TBF5-2 is encoded by the mRNA variant ENST00000296333.10
of PIGX. This isoform has a size of 276 amino acids and a
mass of 30,974 Da, distinguishing it from the canonical sequence at
positions 177-195, which contain the sequence QAGSRRMIRFRFDSFDKTI
(Fig. 3) (2), and comprises the soluble chain in the
lumen of the ER. As for the tertiary structure of PIGX,
bioinformatic predictions in AlphaFold DB of the canonical protein
(19) show that the soluble luminal
amino acid chain in the ER consists of a random coil structure and
β-sheets, while the transmembrane region is a single α-helix
(Fig. 2B). Co-precipitation has
demonstrated that PIGX is associated with PIGM (24). However, whether the transmembrane or
the large luminal domain is implicated in stabilizing the PIGM
protein remains unknown.
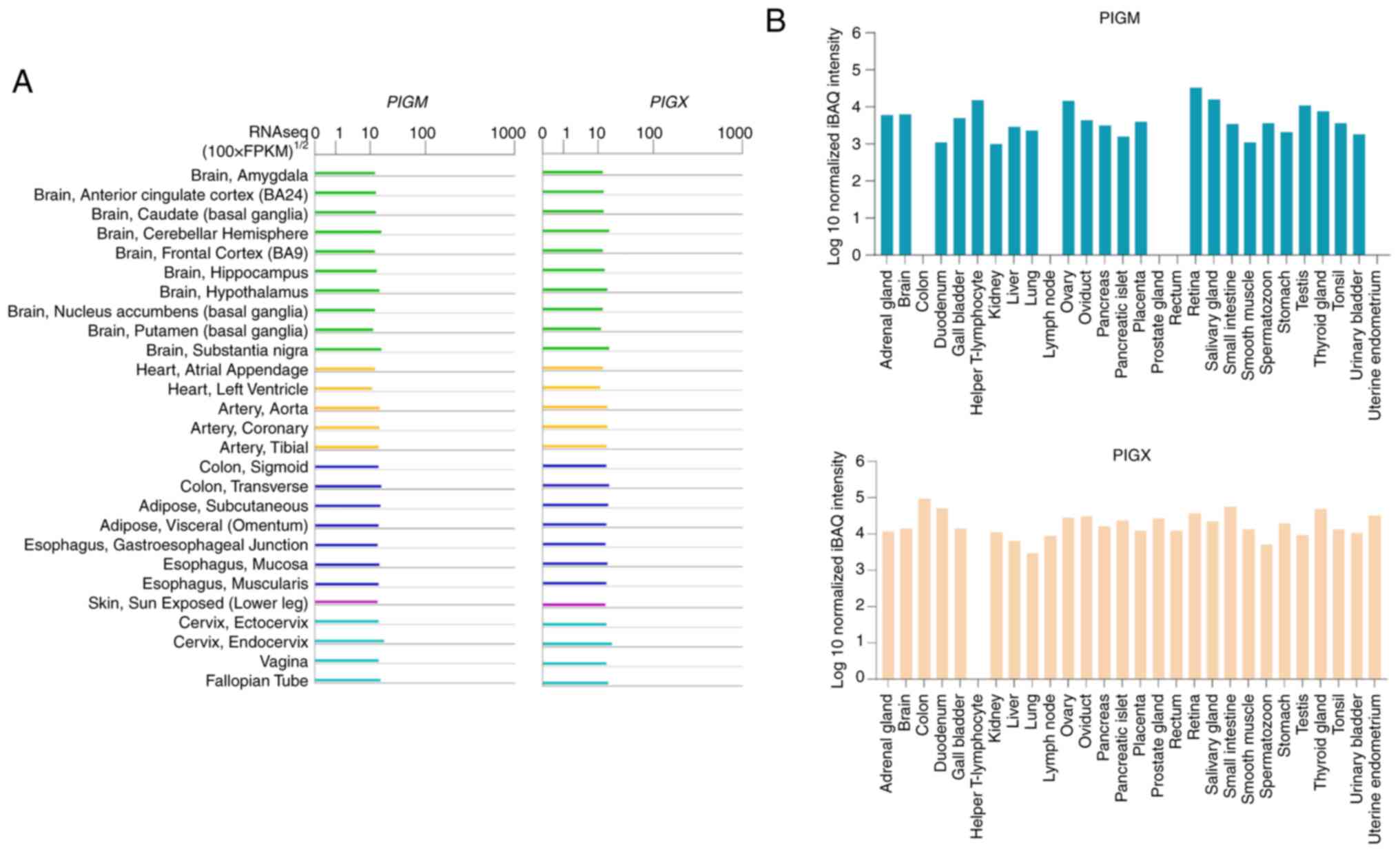
According to the Genotype-Tissue Expression project
(25,26), there is mRNA expression of
PIGM and PIGX in all major tissues (26,27),
including the nervous system, heart, digestive system, skin and
reproductive system in humans (Fig.
4A). Consistently, Proteomics DB reports the expression of both
proteins (PIGX Q8TBF5-1) in the brain and the digestive and
reproductive systems, but also in the breast, lung, retina, kidney
and thyroid gland (Fig. 4B)
(28). Notably, the expression of
PIGM and PIGM has been detected in the colon, T lymphocytes,
prostate and rectum (Fig. 4B)
(28). These data suggest that
GPI-MT-I is present in numerous types of tissues regardless of
their specialized function in humans.
3. Consequences of PIGM and
PIGX knockout (KO)
Mutant mammalian cells with deficiencies in genes
implicated in the GPI-anchor biosynthesis have been previously
reported, including those encoding GPI-GnT and GPI-TA (29), and the genes PIGV,
PIGB, PIGM and PIGX (6,20,29-31).
For example, in vitro experiments conducted using human
lymphoma cells reveal that lack of PIGM results in elevated
GlcN-acyl-PI levels and impaired surface expression of GPI-APs
(Fig. 5) (20). Moreover, a recent study using a KO
human cell library targeting GPI biosynthetic genes indicated that
suppressing expression of specific PIG genes results in the
absence of GPI-APs on the cell surface (6). Notably, KO of regulatory subunits of
GPI-GnT leads to diminished presence of GPI-APs, while KO of
catalytic subunits results in complete absence of these proteins
(Fig. 5). Furthermore, elimination
of GPI-AP synthesis occurs following KO of genes involved in steps
subsequent to GPI-GnT activity. Regarding enzymes catalyzing the
transfer of Man1 and 2, GPI-MT-I (PIGM) and GPI-MT-II
(PIGV), complete removal of GPI-AP presence is observed upon
PIGM KO, whereas residual presence of GPI-APs persists after
PIGX KO (6). Experiments
conducted in CHO cells derived from hamster adult ovaries
demonstrated that defective PIGX leads to the accumulation
of GlcN-acyl-PI, imitating the phenotype observed in
PIGM-deficient cells (32).
Additionally, the aforementioned study revealed diminished
expression of protein PIGM in the absence of PIGX, while a
10-fold increase in expression was observed when PIGX was
expressed. For these reasons, it is hypothesized that PIGX has a
role in stabilizing PIGM (32).
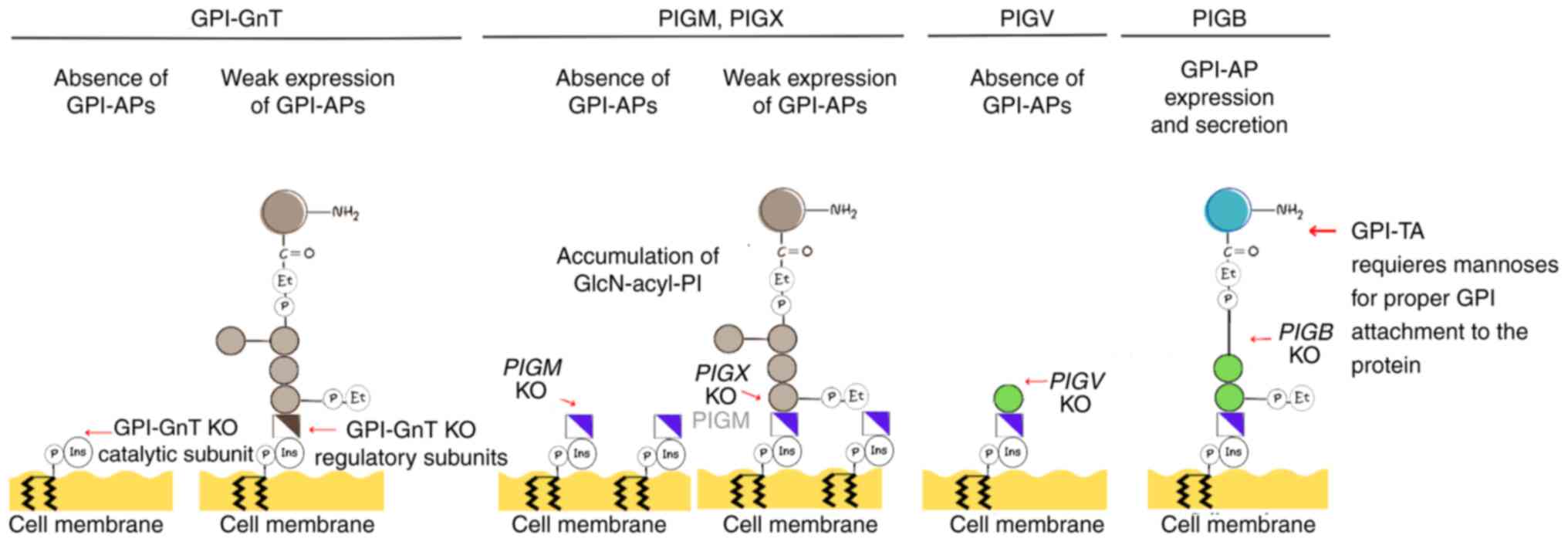 | Figure 5GPI-anchored protein expression
following KO of encoding enzymes in the early stages of GPI
biosynthesis. KO of the catalytic subunits of GPI-GnT produces the
absence of GPI-APs on the cell surface, while knocking out the
regulatory subunits leads to weak expression of the protein.
Knocking out PIGM produces the absence of GPI-APs, whereas
knocking out PIGX leads to low expression of PIGM protein
and weak GPI-AP expression. In both cases, cells display an
accumulation of glucosamine-acyl-phosphatidylinositol. KO of
PIGV leads to the absence of GPI-APs, while the KO of
PIGB does not affect the expression of GPI-APs in the cell
surface or their secretion, suggesting the GPI-TA enzyme requires
three mannoses to attach the GPI core to target proteins. GPI,
glycosylphosphatidylinositol; KO, knock out; GnT,
N-acetylglucosamine transferase; AP, anchored protein; PIG,
phosphatidylinositol-glycan biosynthesis class; TA, transamidase
enzyme complex; Et, ethanolamine; Ins, inositol; P, phosphate. |
Similar outcomes are associated with mutations
affecting other genes involved in GPI synthesis. PIGV KO
completely abolishes the surface expression of GPI-APs in human
cells (Fig. 5). Conversely,
PIGB KO, involved in transferring Man3, allows limited
expression of GPI-APs, while PIGZ KO does not impact GPI-AP
biosynthesis (6). In summary, the
aforementioned studies demonstrate the essential role of enzymes in
the initial stages of GPI synthesis, including GPI-MT-I, for
expression of GPI-APs on the cell surface. Dysregulation of their
expression may lead to alterations in cell surface
characteristics.
4. PIGM and PIGX in human
disease
Paroxysmal nocturnal hemoglobinuria
(PNH)
PNH represents a rare and chronic hematological
disorder resulting from somatic mutations in the X-linked
PIGA gene within hematopoietic stem cells (33). PIGA gene encodes the
catalytic subunit of GPI-GnT (2).
Consequently, hematopoietic stem cells carrying these mutations
give rise to aberrant clone blood cells that lack GPI-APs,
specifically CD55 and CD59. Notably, these proteins serve crucial
roles as regulatory components in the complement system (33). PNH manifests as a hematological
condition marked by intravascular hemolysis, thrombosis and bone
marrow failure, often resulting in cytopenia. The chronic hemolysis
observed in patients with PNH is linked to the absence or
deficiency of GPI-APs (33). This
deficiency disrupts activation of the complement system, leading to
the lysis of immune and red blood cells (33). Small PNH clones with GPI-AP
deficiencies are detected in the bone marrow of patients displaying
PNH-associated symptoms or in healthy individuals (34). An ultra-deep sequencing analysis of
PNH small clones revealed that a patient with classic PNH harbored
a PIGM gene deletion at 459-462, suggesting a protein change
in valine 154, a transmembrane region (2). Despite the absence of reported
PIGX mutations in patients with PNH, there are allele
variants in small PNH clones without clinical relevance (34).
PIGM-associated GPI deficiency
Certain inherited GPI deficiencies are due to
mutations in PIGM. A study of inherited GPI deficiency in
two unrelated consanguineous families characterized by venous
thrombosis and seizures indicated that a hypomorphic promoter
mutation in PIGM causes GPI deficiency (24). Homozygosity mapping demonstrated a
point mutation, 270 C→G, at the promoter of PIGM was
associated with decreased levels of PIGM mRNA. Further
experiments demonstrated that the point mutation disrupted binding
of Sp1, an ubiquitous transcription factor, to a GC box, which is
located proximal to the transcription initiation site; the point
mutation led to decrease in the activity of the PIGM
promoter. Decreased transcription of PIGM led to a blockage
of GPI mannosylation from partial to severe deficiency of GPI
(24). This mutation has also been
described in GPI-inositol deficiency characterized by
cerebrovascular thrombotic events (35). Molecular analysis indicated that
patients were homozygous for the point mutation 270 C→G mutation
and that cells displayed low mRNA expression levels compared with
controls (35).
Further investigation into the mechanism underlying
PIGM deficiency is required to elucidate the mechanism
behind PNH and PIGM deficiency predisposing to thrombosis, a
characteristic that is not observed in other GPI deficiencies
(35). Additionally, patients with
PIGM-associated GPI deficiencies do not display
intravascular hemolysis (35,36).
These phenotypes may be attributed to variations in PIGM
mRNA levels and GPI expression. Specifically, the differential
expression of PIGM occurs in patient-derived B cells
compared with erythrocytes, and it is linked to distinct promoter
chromatin accessibility and binding of Sp1(36).
Cancer
According to gene expression analysis in patients
with cancer, PIGM is upregulated in lung and other types of
cancer, including glioma, skin, liver and thyroid cancer (37). In lung cancer, PIGM, in
combination with other genes, is associated with patient survival
outcomes: Patients with lung cancer and low expression of
PIGM mRNA exhibit higher overall survival than those with
high PIGM mRNA expression (38). Analysis in myeloma showed that high
expression of PIGM is associated with adverse survival
outcomes (37). Expression of
PIGM is notably higher in myeloma samples compared with that
in normal cells. Furthermore, there is a marked increase in
PIGM expression in myeloma cell samples exhibiting
cytogenetic aberrations such as 1q21-gain and 13q14-deletion, and a
corresponding decrease in hyperdiploid myeloma cell samples.
Experimental data in myeloma cells highlight the direct influence
of varying PIGM expression on the presence of the GPI-APs
CD55 and CD59 on the cell surface (37). Analysis of tumor samples revealed
gene alterations in PIGM across either all or most
metastatic sites in ependymoma, with a notable absence of these
alterations at the primary site (39). The aforementioned studies
demonstrate a clear association between malignancy and increased
PIGM expression or genetic modifications in PIGM
gene.
Certain types of cervical cancer display low
expression of PIGX (16),
and mutations in 17 genes, including PIGX, have been
identified in young, non-smoker patients with lung cancer,
displaying potentially pathogenic effects (40). In breast cancer, high levels of
PIGX mRNA are associated with decreased survival in
disease-free patients compared with those with low levels (14). In vitro experiments indicate
that PIGX expression promotes the proliferation of breast
cancer cells. However, PIGM does not affect cell
proliferation, suggesting that PIGX promotes cancer cell
proliferation independently of PIGM (14). Further analysis showed that protein
PIGX may form a protein complex with Reticulocalbin-1 (RCN1) and
RCN2 in the ER, which regulate calcium-dependent activity.
Moreover, in vitro experiments reveal that the silencing of
PIGX, RCN1 or RCN2 results in reduced
expression of the genes Zic family member 1 (ZIC1) and EH
domain containing 2 (EHD2), two putative tumor suppressor
genes (14). PIGX might
contribute to the promotion of cancer cell proliferation by
suppressing EHD2 and ZIC1. Notably, PIGX, in addition
to its association with PIGM, may engage with other proteins in the
ER and exhibit an autonomous functional role (14). It is currently unclear how
PIGX affects the gene expression of EHD2 and
ZIC1.
5. Key roles of PIGM, PIGX and
PIGV in GPI-AP secretion
The role of GPI-MT-I and II in GPI synthesis and
secretion has been studied in hyperphosphatasia with mental
retardation syndrome (HPMRS) (41-44).
This autosomal recessive syndrome is characterized by intellectual
disability and elevated serum and remodeling are associated with
HPMRS, with the PIGV gene being well-studied (41-44).
PIGV mutations include c.53G→A, c.176T→G, c.467G→A,
c.905T→C, c.1022C→A and c.1405C→T and in some instances, mutations
can be biallelic (42,43). Experiments using PIGV with
transmembrane region mutations suggest that these mutations may
destabilize the protein (41).
Cells deficient in PIGV and PIGB
secrete GPI-APs into the medium, accompanied by the accumulation of
incomplete GPI-bearing Man (41).
This phenotype is explained by the fact that secretion of alkaline
phosphatase (ALP) requires GPI-TA, which, in normal cells, cleaves
the C-terminal GPI attachment signal peptide and replaces it with
GPI. In PIGX-deficient cells, where incomplete shorter GPIs
lacking Man accumulate, ALP is degraded. This suggests that at
least one Man residue is required for GPI-TA to cleave the GPI
attachment signal. Consequently, it is hypothesized that GPI-TA
recognizes incomplete GPI-bearing mannose, cleaving a hydrophobic
signal peptide, resulting in secretion of soluble ALP (Fig. 5) (41). Although HPMRS is not observed in
patients with a PIGM mutation (41), the aforementioned study suggested a
key step during GPI synthesis involving the enzymes GPI-MT-I
(PIGM), PIGX and GPI-MT-II (PIGV) for proper
recognition of GPI-APs by GPI-TA and subsequent secretion.
Studies indicated a crucial role of GPI-TA in
regulating glycolipid biosynthesis, since GPI biosynthesis is
suppressed by the ER-associated degradation pathway when cells are
defective in transferring the complete GPI core to proteins to
prevent GPI accumulation (9,45).
Biosynthesis of GPI is upregulated in ER-associated
degradation-deficient cells (46).
Whether the ER-associated degradation pathway is triggered in
PIGM-deficient cells or in cells with decreased expression
of PIGM or PIGX that accumulate GlcN-acyl-PI remains
unclear.
6. Interaction between PIGM and PIGX and
other proteins
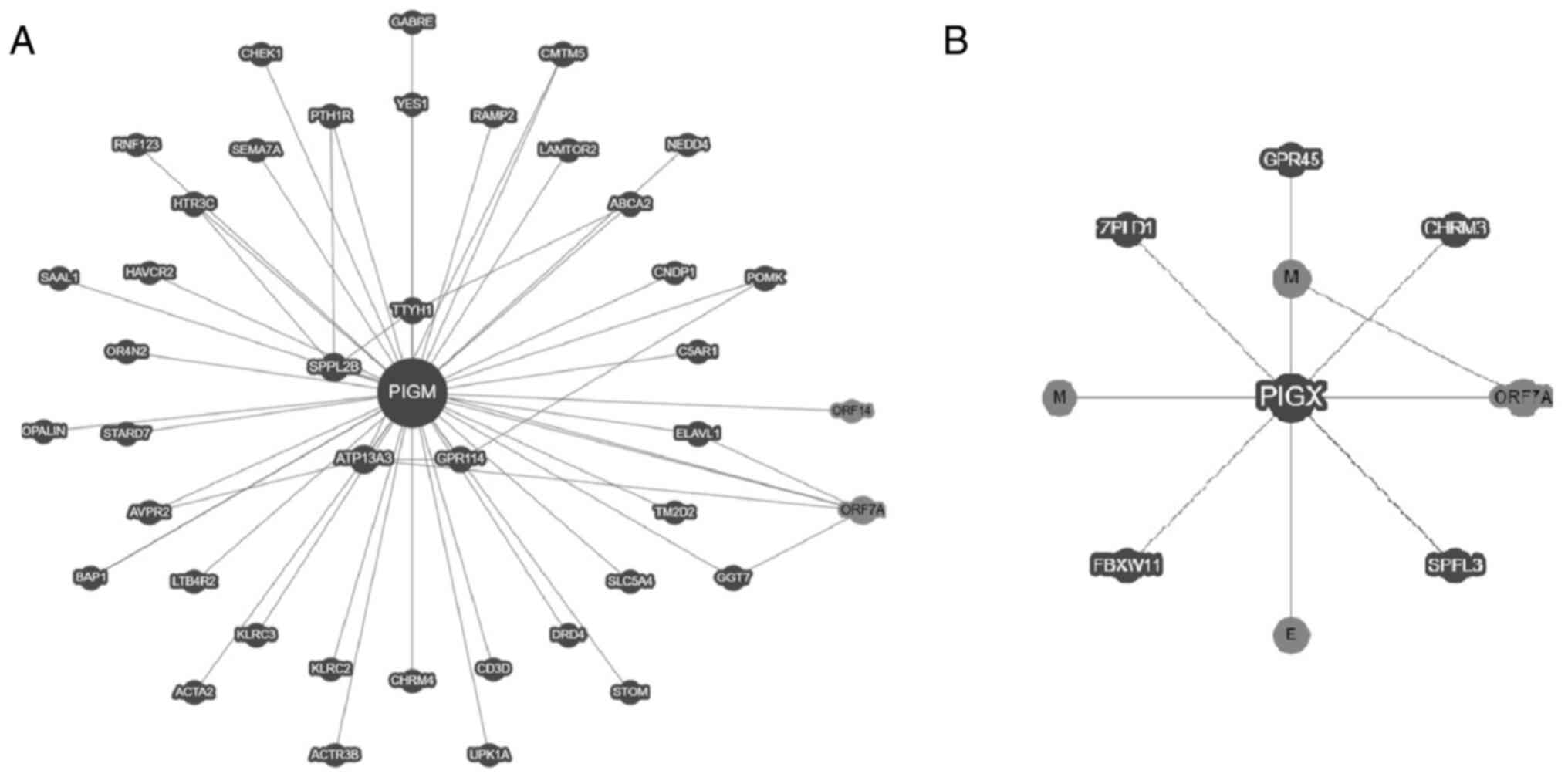
Several studies have investigated the human
interactome and its implication in human diseases (47-49).
BioGRID, a public database of genetic and protein interactions
(23), indicated that PIGM and PIGX
proteins interact physically with other proteins (Fig. 6). Affinity capture-mass spectrometry
and two-hybrid assays indicate that PIGM can bind to 39 proteins
(Fig. 6A) and PIGX can bind to six
(Fig. 6B). Notably, each subunit
binds to different proteins. Proteins interacting with PIGM include
glycoproteins, G-protein-coupled receptors in humans and the
proteins ORF7, ORF14, E and M of severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) (Fig.
6A) (23). Whether these
interactions reveal a specific biological context of ER or a
cross-regulation of the proteins remains unknown; G-protein-coupled
receptors, glycoproteins and GPI-APs are commonly recruited in
lipid rafts (50-52)
and recent evidence indicates that GPI-biosynthesis is relevant for
the life cycle of SARS-CoV-2(53).
7. Conclusion
In summary, expression of PIGM and
PIGX is reported in most types of cell, suggesting that
GPI-APs may be present in most cells. The present review indicated
that PIGM and PIGX are key genes for GPI synthesis
since their absence may lead to accumulation of GPIs that lack Man
in the cell and deficiency of GPI-APs in the cell membrane or their
secretion. Absence or altered expression of PIGM gene is
associated with PNH and inherited GPI deficiency, which are
characterized by thrombosis; whether this phenotype is due to
altered expression of specific GPI-APs remains unclear. Altered
expression of PIGM and PIGX has been reported in
cancer (14,16,37,39);
to the best of our knowledge, however, whether these changes may
lead to altered expression of GPI-APs that drive malignant
phenotype has not been explored. Transcription factor Sp1 may exert
a role in the transcription of PIGM; to the best of our
knowledge, there are no studies regarding the transcription of
PIGX. Regulation of GPI-MT-I enzyme activity depends on the
expression of PIGX and, to the best of our knowledge, no
other mechanism has been described. However, since PIGM and PIGX
may interact with other proteins independently, additional
regulation may be involved. Further exploration may enable the
development of targeted therapies for cancer.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
ATV performed the literature review and wrote the
manuscript. VVR wrote and reviewed the manuscript. LMF and PMM
performed the literature review, supervised the study and wrote and
reviewed the manuscript. Data authentication is not applicable. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kinoshita T: Biosynthesis and biology of
mammalian GPI-anchored proteins. Open Biol.
10(190290)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Coudert E, Gehant S, De Castro E, Pozzato
M, Baratin D, Neto T, Sigrist CJ, Redaschi N and Bridge A: UniProt
Consortium. Annotation of biologically relevant ligands in
UniProtKB using ChEBI. Bioinformatics. 39(btac793)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wu T, Yin F, Guang S, He F, Yang L and
Peng J: The glycosylphosphatidylinositol biosynthesis pathway in
human diseases. Orphanet J Rare Dis. 15(129)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Komath SS, Fujita M, Hart GW, Ferguson MAJ
and Kinoshita T: Glycosylphosphatidylinositol anchors. In: Varki A,
Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Mohnen D,
Kinoshita T, Packer NH, Prestegard JH (eds) et al:
Essentials of Glycobiology [Internet]. 4th edition. Cold Spring
Harbor (NY): Cold Spring Harbor Laboratory Press: Chapter 12, 2022.
Available from: https://www.ncbi.nlm.nih.gov/books/NBK579963/.
|
|
5
|
Liu YS and Fujita M: Mammalian GPI-anchor
modifications and the enzymes involved. Biochem Soc Trans.
48:1129–1138. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liu SS, Liu YS, Guo XY, Murakami Y, Yang
G, Gao XD, Kinoshita T and Fujita M: A knockout cell library of GPI
biosynthetic genes for functional studies of GPI-anchored proteins.
Commun Biol. 4(777)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yadav U and Khan MA: Targeting the GPI
biosynthetic pathway. Pathog Glob Health. 112:115–122.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lopez S, Rodriguez-Gallardo S, Sabido-Bozo
S and Muñiz M: Endoplasmic reticulum export of GPI-anchored
proteins. Int J Mol Sci. 20(3506)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang Y, Maeda Y, Liu YS, Takada Y,
Ninomiya A, Hirata T, Fujita M, Murakami Y and Kinoshita T:
Cross-talks of glycosylphosphatidylinositol biosynthesis with
glycosphingolipid biosynthesis and ER-associated degradation. Nat
Commun. 11(860)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hirata T, Mishra SK, Nakamura S, Saito K,
Motooka D, Takada Y, Kanazawa N, Murakami Y, Maeda Y, Fujita M, et
al: Identification of a Golgi GPI-N-acetylgalactosamine transferase
with tandem transmembrane regions in the catalytic domain. Nat
Commun. 9(405)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lebreton S, Zurzolo C and Paladino S:
Organization of GPI-anchored proteins at the cell surface and its
physiopathological relevance. Crit Rev Biochem Mol Biol.
53:403–419. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bellai-Dussault K, Nguyen TTM, Baratang
NV, Jimenez-Cruz DA and Campeau PM: Clinical variability in
inherited glycosylphosphatidylinositol deficiency disorders. Clin
Genet. 95:112–121. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Paprocka J, Hutny M, Hofman J, Tokarska A,
Kłaniewska M, Szczałuba K, Stembalska A, Jezela-Stanek A and
Śmigiel R: Spectrum of neurological symptoms in
glycosylphosphatidylinositol biosynthesis defects: Systematic
review. Front Neurol. 12(758899)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nakakido M, Tamura K, Chung S, Ueda K,
Fujii R, Kiyotani K and Nakamura Y: Phosphatidylinositol glycan
anchor biosynthesis, class X containing complex promotes cancer
cell proliferation through suppression of EHD2 and ZIC1, putative
tumor suppressors. Int J Oncol. 49:868–876. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cao J, Wang P, Chen J and He X: PIGU
overexpression adds value to TNM staging in the prognostic
stratification of patients with hepatocellular carcinoma. Hum
Pathol. 83:90–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Martinez-Morales P, Morán Cruz I, Roa-de
la Cruz L, Maycotte P, Reyes Salinas JS, Vazquez Zamora VJ,
Gutierrez Quiroz CT, Montiel-Jarquin AJ and Vallejo-Ruiz V:
Hallmarks of glycogene expression and glycosylation pathways in
squamous and adenocarcinoma cervical cancer. PeerJ.
9(e12081)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Baratang NV, Jimenez Cruz DA, Ajeawung NF,
Nguyen TTM, Pacheco-Cuéllar G and Campeau PM: Inherited
glycophosphatidylinositol deficiency variant database and analysis
of pathogenic variants. Mol Genet Genomic Med.
7(e00743)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cunningham F, Allen JE, Allen J,
Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov
AG, Barnes I, Bennett R, et al: Ensembl 2022. Nucleic Acids Res. 50
(D1):D988–D995. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jumper J, Evans R, Pritzel A, Green T,
Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A,
Potapenko A, et al: Highly accurate protein structure prediction
with AlphaFold. Nature. 596:583–589. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Maeda Y, Watanabe R, Harris CL, Hong Y,
Ohishi K, Kinoshita K and Kinoshita T: PIG-M transfers the first
mannose to glycosylphosphatidylinositol on the lumenal side of the
ER. EMBO J. 20:250–261. 2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sayers EW, Bolton EE, Brister JR, Canese
K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, et al:
Database resources of the national center for biotechnology
information. Nucleic Acids Res. 50 UD1):D20–D26. 2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kearse MG and Wilusz JE: Non-AUG
translation: A new start for protein synthesis in eukaryotes. Genes
Dev. 31:1717–1731. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Oughtred R, Rust J, Chang C, Breitkreutz
BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, et
al: The BioGRID database: A comprehensive biomedical resource of
curated protein, genetic, and chemical interactions. Protein Sci.
30:187–200. 2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Almeida AM, Murakami Y, Layton DM, Hillmen
P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I,
Mollica L, et al: Hypomorphic promoter mutation in PIGM causes
inherited glycosylphosphatidylinositol deficiency. Nat Med.
12:846–851. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
GTEx Consortium: The genotype-tissue
expression (GTEx) project. Nat Genet. 45:580–585. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
GTEx Portal: Available in: gtexportal.org/home/gene/PIGX. Reviewed on
05/07/2023.
|
|
27
|
Stelzer G, Rosen R, Plaschkes I, Zimmerman
S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et
al: The GeneCards suite: From gene data mining to disease genome
sequence analyses. Curr Protoc Bioinformatics. 54:1.30.1–1.30.33.
2016.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Samaras P, Schmidt T, Frejno M, Gessulat
S, Reinecke M, Jarzab A, Zecha J, Mergner J, Giansanti P, Ehrlich
HC, et al: ProteomicsDB: A multi-omics and multi-organism resource
for life science research. Nucleic Acids Res. 48 (D1):D1153–D1163.
2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Maeda Y, Ashida H and Kinoshita T: CHO
glycosylation mutants: GPI anchor. Methods Enzymol. 416:182–205.
2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hyman R: Somatic genetic analysis of the
expression of cell surface molecules. Trends Genet. 4:5–8.
1988.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kang JY, Hong Y, Ashida H, Shishioh N,
Murakami Y, Morita YS, Maeda Y and Kinoshita T: PIG-V involved in
transferring the second mannose in glycosylphosphatidylinositol. J
Biol Chem. 280:9489–9497. 2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ashida H, Hong Y, Murakami Y, Shishioh N,
Sugimoto N, Kim YU, Maeda Y and Kinoshita T: Mammalian PIG-X and
yeast Pbn1p are the essential components of
glycosylphosphatidylinositol-mannosyltransferase I. Mol Biol Cell.
16:1439–1448. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Bektas M, Copley-Merriman C, Khan S, Sarda
SP and Shammo JM: Paroxysmal nocturnal hemoglobinuria: Role of the
complement system, pathogenesis, and pathophysiology. J Manag Care
Spec Pharm. 26 (12-b Suppl):S3–S8. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jeong D, Park HS, Kim SM, Im K, Yun J, Lee
YE, Ryu S, Ahn YO, Yoon SS and Lee DS: Ultradeep sequencing
analysis of paroxysmal nocturnal hemoglobinuria clones detected by
flow cytometry: PIG mutation in small PNH clones. Am J Clin Pathol.
156:72–85. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pode-Shakked B, Heimer G, Vilboux T,
Marek-Yagel D, Ben-Zeev B, Davids M, Ferreira CR, Philosoph AM,
Veber A, Pode-Shakked N, et al: Cerebral and portal vein
thrombosis, macrocephaly and atypical absence seizures in
glycosylphosphatidyl inositol deficiency due to a PIGM promoter
mutation. Mol Genet Metab. 128:151–161. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Costa JR, Caputo VS, Makarona K, Layton
DM, Roberts IAG, Almeida AM and Karadimitris A: Cell-type-specific
transcriptional regulation of PIGM underpins the divergent
hematologic phenotype in inherited GPl deficiency. Blood.
124:3151–3154. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Moehler TM, Seckinger A, Hose D, Andrulis
M, Moreaux J, Hielscher T, Willhauck-Fleckenstein M, Merling A,
Bertsch U, Jauch A, et al: The glycome of normal and malignant
plasma cells. PLoS One. 8(e83719)2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sayeeram D, Katte TV, Bhatia S, Jai Kumar
A, Kumar A, Jayashree G, Rachana DS, Nalla Reddy HV, Arvind
Rasalkar A, Malempati RL and Reddy S DN: Identification of
potential biomarkers for lung adenocarcinoma. Heliyon.
6(e05452)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pisapia DJ, Salvatore S, Pauli C, Hissong
E, Eng K, Prandi D, Sailer VW, Robinson BD, Park K, Cyrta J, et al:
Next-generation rapid autopsies enable tumor evolution tracking and
generation of preclinical models. JCO Precis Oncol.
2017(PO.16.00038)2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fu F, Tao X, Jiang Z, Gao Z, Zhao Y, Li Y,
Hu H, Shen L, Sun Y and Zhang Y: Identification of germline
mutations in east-asian young never-smokers with lung
adenocarcinoma by whole-exome sequencing. Phenomics. 3:182–189.
2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Murakami Y, Kanazawa N, Saito K, Krawitz
PM, Mundlos S, Robinson PN, Karadimitris A, Maeda Y and Kinoshita
T: Mechanism for release of alkaline phosphatase caused by
glycosylphosphatidylinositol deficiency in patients with
hyperphosphatasia mental retardation syndrome. J Biol Chem.
287:6318–6325. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Horn D, Krawitz P, Mannhardt A, Korenke GC
and Meinecke P: Hyperphosphatasia-mental retardation syndrome due
to PIGV mutations: Expanded clinical spectrum. Am J Med Genet A.
155A:1917–1922. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Horn D, Wieczorek D, Metcalfe K, Barić I,
Paležac L, Cuk M, Petković Ramadža D, Krüger U, Demuth S, Heinritz
W, et al: Delineation of PIGV mutation spectrum and associated
phenotypes in hyperphosphatasia with mental retardation syndrome.
Eur J Hum Genet. 22:762–767. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Fu L, Liu Y, Chen Y, Yuan Y and Wei W:
Mutations in the PIGW gene associated with hyperphosphatasia and
mental retardation syndrome: A case report. BMC Pediatrics.
19(68)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Aguilera-Romero A and Muñiz M: GPI
anchors: Regulated as needed. J Cell Biol.
222(e202303097)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Liu YS, Wang Y, Zhou X, Zhang L, Yang G,
Gao XD, Murakami Y, Fujita M and Kinoshita T: Accumulated
precursors of specific GPI-anchored proteins upregulate GPI
biosynthesis with ARV1. J Cell Biol. 222(e202208159)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang J, Huo K, Ma L, Tang L, Li D, Huang
X, Yuan Y, Li C, Wang W, Guan W, et al: Toward an understanding of
the protein interaction network of the human liver. Mol Syst Biol.
13(965)2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Huttlin EL, Ting L, Bruckner RJ, Gebreab
F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al:
The BioPlex network: A systematic exploration of the human
interactome. Cell. 162:425–440. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Huttlin EL, Bruckner RJ, Paulo JA, Cannon
JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et
al: Architecture of the human interactome defines protein
communities and disease networks. Nature. 545:505–509.
2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Brown DA and Jacobson K: Microdomains,
lipid rafts and caveolae (San Feliu de Guixols, Spain, 19-24 May
2001). Traffic. 2:668–672. 2001.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Galbiati F, Ranani B and Lisanti MP:
Emerging themes in lipid rafts and caveolae. Cell. 106:403–411.
2001.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Suzuki KG, Fujiwara TK, Sanematsu F, Iino
R, Edidin M and Kusumi A: GPI-anchored receptor clusters
transiently recruit Lyn and G alpha for temporary cluster
immobilization and Lyn activation: Single-molecule tracking study
1. J Cell Biol. 177:717–730. 2007.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Schneider WM, Luna JM, Hoffmann HH,
Sánchez-Rivera FJ, Leal AA, Ashbrook AW, Le Pen J, Ricardo-Lax I,
Michailidis E, Peace A, et al: Genome-scale identification of
SARS-CoV-2 and pan-coronavirus host factor networks. Cell.
184:120–132.e14. 2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Sievers F, Wilm A, Dineen DG, Gibson TJ,
Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al:
Fast, scalable generation of high-quality protein multiple sequence
alignments using Clustal Omega. Mol Syst Biol.
7(539)2011.PubMed/NCBI View Article : Google Scholar
|