Introduction
Chronic kidney disease (CKD) is a long-term
condition that causes progressive damage to kidney parenchyma,
leading to the deterioration of kidney function and gradually
progressing to end-stage kidney disease (ESKD) (1). ESKD is life-threatening without kidney
replacement treatment, such as dialysis and kidney transplantation.
Peritoneal dialysis (PD) uses the peritoneum in the patient's
abdomen as the membrane through which the dialysate passes, and via
this process, fluid and dissolved substances are exchanged with the
blood. The excess fluid, toxins and other substances that result
from kidney failure are also removed through this process (2). PD has the advantage that it can be
performed at home, thereby obviating the need for the patient to be
admitted to hospital; moreover, it is more cost-effective, is
associated with fewer dietary restrictions, and increases both the
perception of freedom for patients and patient satisfaction,
thereby improving the patient's quality of life (3). In spite of these benefits, however, PD
still carries a high risk of peritoneal infection, subcutaneous
tunnel infection and catheter exit site infection (4-6).
However, even though this dialysis technique has a number of
benefits when compared with other available methods, there is an
increased risk of peritonitis and microbial contamination of the
blood through the catheter which compromises the immune defense
system of patients, leading to complications, morbidity and
mortality (3,4).
Currently, next-generation sequencing (NGS) provides
a promising alternative approach for broad microbial identification
in clinical samples (7,8). This approach allows for the unbiased
detection of almost all potential microorganisms, including
bacteria, fungi, viruses and parasites. The NGS approach can be
used to overcome the limitations of traditional methods, whereby
the presence of microorganisms can be detected through the unique
classification of DNA/RNA sequences (7). This approach has been successfully
applied in the clinical diagnosis of infectious diseases such as
severe acute respiratory syndrome coronavirus (9), nosocomial infection detection
(10), Clostridium
difficile-associated disease (11), as well as in response to outbreaks
of disease and the discovery of novel pathogens (7,12).
Particularly purulent fluids are often indicative of an infectious
etiology, and application of the NGS approach has the potential to
decrease the assay sensitivity and shorten time detection (13). The majority of metagenomic studies
have used Illumina sequencing platforms, with sequencing turnaround
times exceeding 16 h and overall process turnaround times of 48-72
h. By contrast, the company Oxford Nanopore Technologies (ONT)
offers a third-generation sequencing technology that has several
advantages compared with second-generation technologies in terms of
longer reads and real-time sequence analysis capabilities, allowing
the detection of potential microorganisms within hours of
sequencing, and requiring shorter turnaround times of <6 h
(13,14).
However, the precise nature of the microbial
contamination that may influence the occurrence of infection
remains unclear, and this represents a gap in our knowledge that
urgently needs to be filled. Few studies have used NGS techniques
to explore the microbial associated with PD-related peritonitis
through short-reads sequencing platform; however, the taxonomic
resolution is limited at the genus level (15,16).
Therefore, the present study aimed to develop the rapid diagnosis
pipeline for bacterial species classification in the PD effluent
(PDE) of patients with ESKD during an early phase of infection
based on 16S ribosomal DNA (rDNA) long-reads amplicon sequencing.
Furthermore, the accuracy of the technique was comparable with the
gold standard culture-dependent method. Thus, the present study was
conducted to utilize the advantages of ONT in discovery and
characterization of the PD-related microbial infection along with a
comparison with the culture-dependent approach.
Patients and methods
Participants
The present study utilized 104 PDE samples obtained
from patients (62 men and 42 women; age, 57.88±14.43 years) at the
Thailand-Peritoneal Dialysis Outcomes and Practice Patterns Study
(PDOPPS) facility of 22 hospitals in Thailand between January 2019
and December 2021. The PDOPPS was approved by the Institutional
Review Board of the Faculty of Medicine, Chulalongkorn University
(approval no. 1544/2020; IRB no. 499/58; Bangkok, Thailand). The
present study was performed with remains of previously obtained
samples from the PDOPPS, which was approved by the Institutional
Review Board of the Faculty of Medicine, Chulalongkorn University
(COA no. 0754/2022; IRB no. 0253/65; Bangkok, Thailand). Informed
consent was obtained from all included patients.
All participants were >18 years of age, and were
diagnosed with kidney failure, having undergone continuous
ambulatory peritoneal dialysis or automated peritoneal dialysis for
at least 1 month. Peritonitis was diagnosed according to the
presence of two out of three of the following inclusion criteria:
i) PDE from the first episode of infection was observed to be
cloudy, ii) peritonitis caused by the infection had been reported
and iii) the patient had a white blood cell count of >100 cells
and/or the percentage of polymorphonuclear neutrophils was >50%.
By contrast, patients <18 years of age, and those undergoing
hemodialysis, acute peritoneal dialysis or combination renal
replacement therapy were excluded from participation in the present
study.
Sample processing and DNA
extraction
Briefly, the collected samples (50 ml) were
subjected to centrifugation at 25˚C for 15 min in a microcentrifuge
at 18,500 x g, with subsequent removal of the supernatant. The
resultant pellet was suspended in 400 µl sorbitol buffer containing
50 U Lyticase enzyme (Merck KGaA), and then incubated at 30˚C for
60 min to break the cells. The cell suspension was then centrifuged
at 18,500 x g at 4˚C for 5 min, followed by discarding of the
supernatant. Subsequently, 567 µl 1M TE buffer, 3 µl 10% SDS (Merck
KGaA), 100 µl 10 mg/ml lysozyme enzyme (Merck KGaA) and 5 µl 20
mg/ml Proteinase K (Worthington Biochemical Corporation) were added
to the solution. The resultant mixture was incubated at 65˚C for 90
min to digest the proteins and to inhibit the RNases that were
present. Finally, the samples were subjected to centrifugation in a
microcentrifuge at 18,500 x g at 4˚C for 5 min, and the resultant
pellets were collected and resuspended in 200 µl sterile water.
Total genomic DNA (gDNA) extraction was performed using the
magLEAD® 12gC system (Precision System Science Co.,
Ltd.), following the manufacturer's protocol. The quantity and
quality of the DNA were assessed using a 260/280 spectrophotometry
ratio as a measure of DNA purity (1.70-1.85) and 1.5% agarose gel
electrophoresis with RedSafe™ Nucleic Acid Staining Solution (cat.
no. 21141; Intron Biotechnology, Inc.), respectively, and the
extracted DNA was stored at 20˚C until further use.
Bacterial culture
Three 50 ml PDE bags were centrifuged (3,500 x g) at
25˚C for 15 min, and the supernatants were discarded. Of the
remaining solution, 5 ml was mixed into the pellet and injected
into BACTEC™ Plus Aerobic/F and BACTEC Plus Anaerobic/F vials
(Becton, Dickinson and Company). The mixture was subsequently
spread onto various agar plates, including blood agar,
Oxoid® MacConkey agar and chocolate agar (Thermo Fisher
Scientific, Inc.), and thiosulfate citrate bile salt sucrose agar
(Biomedia Thailand Co., Ltd.), as required. The plates were then
incubated at 37˚C for 5-7 days to facilitate bacterial culture.
Identification of bacterial pathogens was subsequently performed
via Gram staining, utilizing the VITEK® MS system
(BIOMÉRIEUX).
16S rDNA gene amplification
PCR amplification of partial 16S rDNA (the V1-V4
region) was selected as the method to classify bacterial
communities at the species level. The primers included specific
target primer sequences (shown underlined) and nanopore adaptor
tails, as follows: 16S_27 forward, 5'
-TTTCTGTTGGTGCTGATATTGCAGRGTTYGATYMTGGCTCAG-3'; and 16S_806
reverse, 5'-ACTTGCCTGTCGCTCTATCTTCGGACTACHVGGGTWTCTAAT-3'. The
representative PCR products (~800 bp) obtained from the
amplification of the 16S rDNA gene using 16S_27F/16S_806R primers
are shown in Fig. S1. The PCR
products were amplified using Phusion™ Plus DNA Polymerase (Thermo
Fisher Scientific, Inc.) to minimize errors during amplification.
The PCR reactions were performed in a total volume of 20 µl,
containing 10 µM forward and reverse primer pairs, 10 µM dNTPs, 0.4
U Phusion™ Plus DNA Polymerase and 10 ng DNA template. The
thermocycling conditions were as follows: 98˚C for 30 sec, 25
cycles of 98˚C for 10 sec, 60˚C for 10 sec, 72˚C for 50 sec, and a
final extension at 72˚C for 5 min. The DNA library of each sample
was pooled using the PCR Barcoding Expansion 1-96 kit (cat. no.
EXP-PBC096; Oxford Nanopore Technologies). The barcoding step made
use of a thermal profile similar to the first PCR, albeit with the
barcode primers instead of primer-specific 16S rDNA genes. The
barcoding step comprised the following thermocycling conditions:
98˚C for 30 sec, 25 cycles of 98˚C for 10 sec, 60˚C for 10 sec,
72˚C for 50 sec and a final extension at 72˚C for 5 min. The PCR
products were subsequently separated using 1% agarose gel
electrophoresis with RedSafe™ Nucleic Acid Staining Solution and
purified using a QIAquick Gel Extraction Kit (cat. no. 28704;
Qiagen GmbH), following the manufacturer's protocol.
Nanopore library preparation and
sequencing
The DNA libraries were quantified using Quant-iT™
dsDNA High Sensitivity Assay Kits (cat. no. Q32851) for the Qubit™
4 Fluorometer (Thermo Fisher Scientific, Inc.). Subsequently, all
libraries were pooled (final concentration 1 µg) for multiplexing,
and underwent purification using 0.5X Agencourt AMPure XP beads
(Beckman Coulter, Inc.). Following purification, the Ligation
Sequencing Kit of Oxford Nanopore Technologies (cat. no.
SQK-LSK112) was used to repair and ligate the ends of the DNA
library. Finally, the pooled DNA library was sequenced (single-end,
800 bp) using a MinION™ Mk1C sequencing device and an R10.4 flow
cell (cat. no. FLO-MIN112), both from Oxford Nanopore
Technologies.
Data analysis
The raw data or FAST5 data were base-called using
Guppy base-caller version 6.0.7 (Oxford Nanopore Technologies) with
a super-accuracy model to generate pass reads in FASTQ format with
a minimum acceptable quality score of Q>10(17). Subsequently, MinIONQC was used to
assess the quality of reads for nanopore data (18). The FASTQ sequences underwent
demultiplexing and adaptor-trimming using Porechop, version 0.2.4.
Filtered reads were clustered, polished and taxonomically
classified using NanoCLUST (19).
The classification was based on the V1-V4 region of 16S rDNA gene
sequences from the Ribosomal Database Project database (20). Relative abundance and taxonomic
assignment data were converted into the QIIME2 data format,
demonstrating bacterial species' richness and evenness based on
taxa abundances, and visualized using GraphPad Prism 9.5.0. This
analysis utilized a plug-in implemented for QIIME2 software
version, 2021.2(21). The
unsupervised clustering was conducted and represented in a heatmap
using the ComplexHeatmap R package (22).
Results
Diversity of bacteria in PDE
The bacterial 16S rDNA (V1-V4 variable region) was
sequenced using a high-throughput MinION™ platform (Oxford Nanopore
Technologies). A total of 1,341,989 raw reads were obtained in the
present study, with 15,079±11,214 average reads/sample. The average
number of classified reads was 10,656±7,762 reads/sample. A
rarefaction analysis was subsequently applied to estimate whether
there was sufficient sequence coverage both to classify all samples
reliably from the bacterial taxa, and to classify them into
operational taxonomic units. The results revealed sufficient
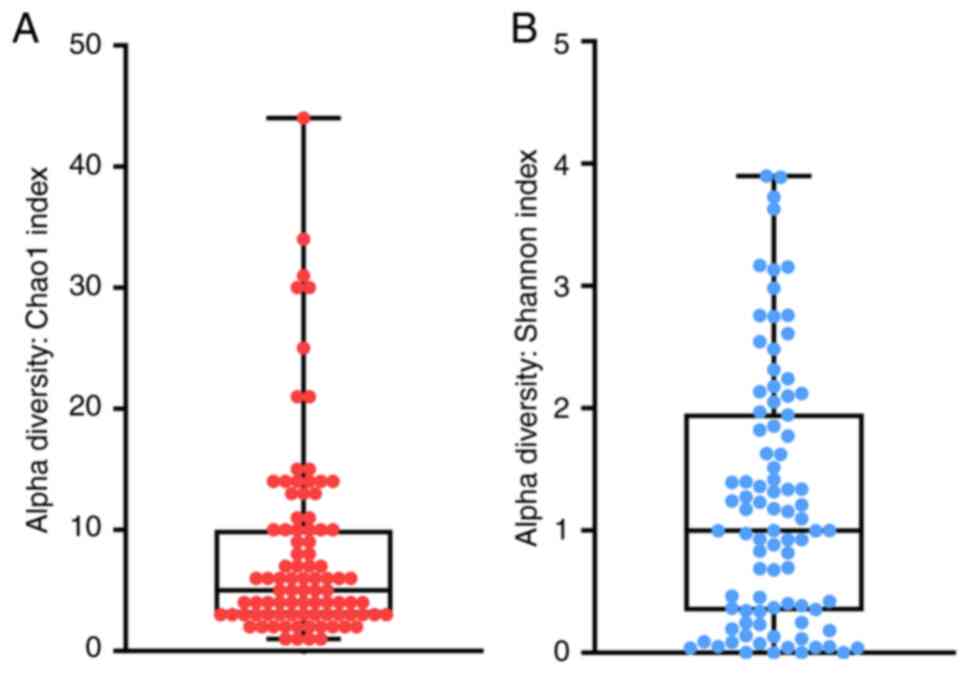
sequencing depth for diversity in 89 PDE samples (Fig. S2). The alpha diversity was assessed
based on Chao1 and Shannon indexes, as shown in Fig. 1A and B. The Chao1 index (8.15±8.06) indicated
the richness of bacteria in each sample, whereas the Shannon index
(1.24±1.05) indicated the richness and evenness of bacteria in each
sample. This result demonstrated that the samples were highly
heterogeneous, which suggests that differences in patient hygiene
may account for the variety of bacterial diversity amongst
patients.
Relative bacterial abundance in PDE of
patients with ESKD
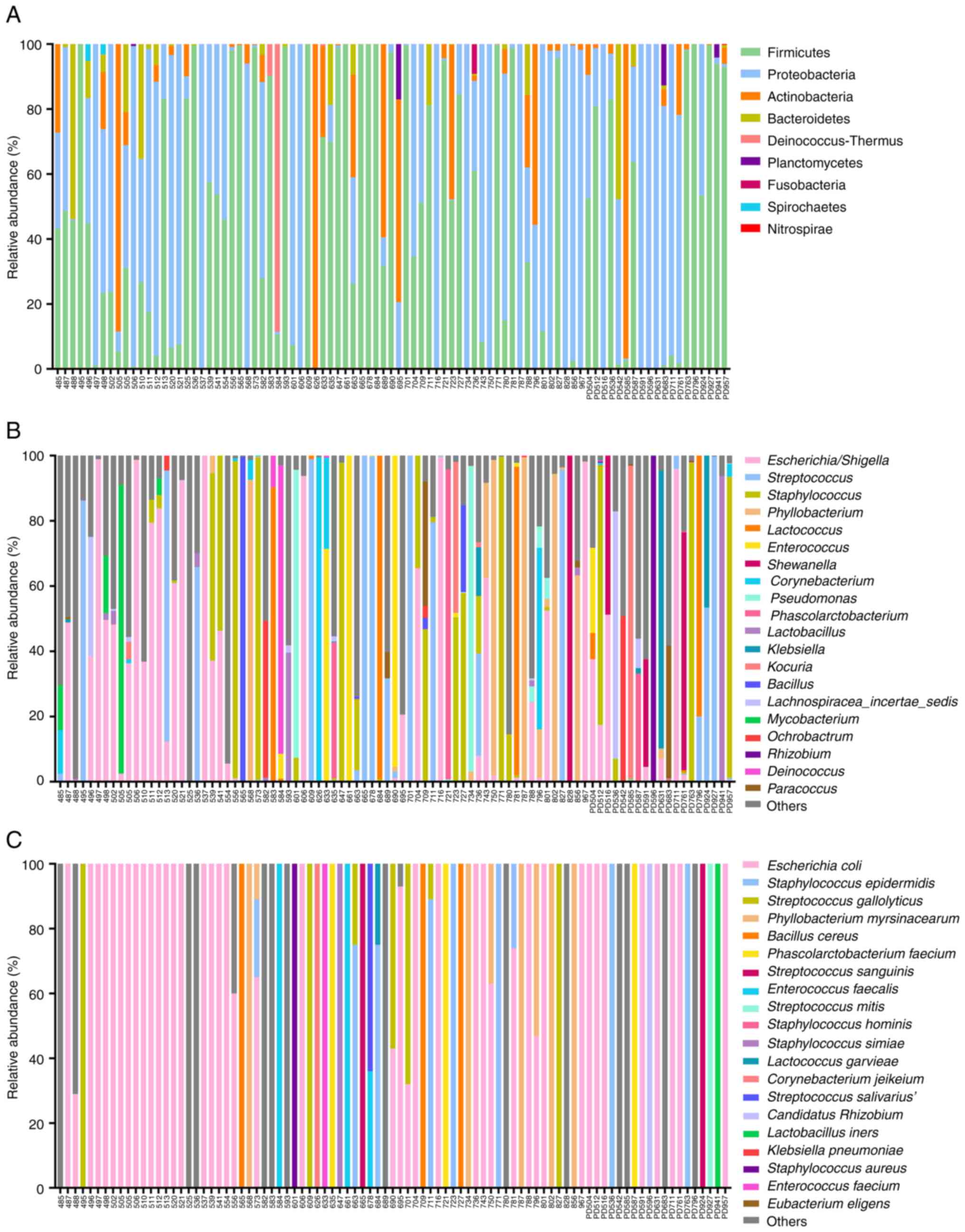
The relative abundance of bacterial composition in
89 PDE samples was classified. At the phylum level, the dominant
bacteria were Firmicutes, Proteobacteria and Actinobacteria
(Fig. 2A). The relative abundance
of bacteria at the genus level is illustrated in Fig. 2B. The five major bacterial genera
were found to be Escherichia/Shigella, Streptococcus,
Staphylococcus, Phyllobacterium and
Lactococcus. Several abundant bacterial species were
identified in patients receiving PD (Fig. 2C). The results showed that
Escherichia coli (E. coli) was the most abundant
bacterial species, followed (in order) by Phyllobacterium
myrsinacearum (P. myrsinacearum), Streptococcus
gallolyticus (S. gallolyticus), Staphylococcus
epidermidis (S. epidermidis) and Shewanella algae
(S. algae).
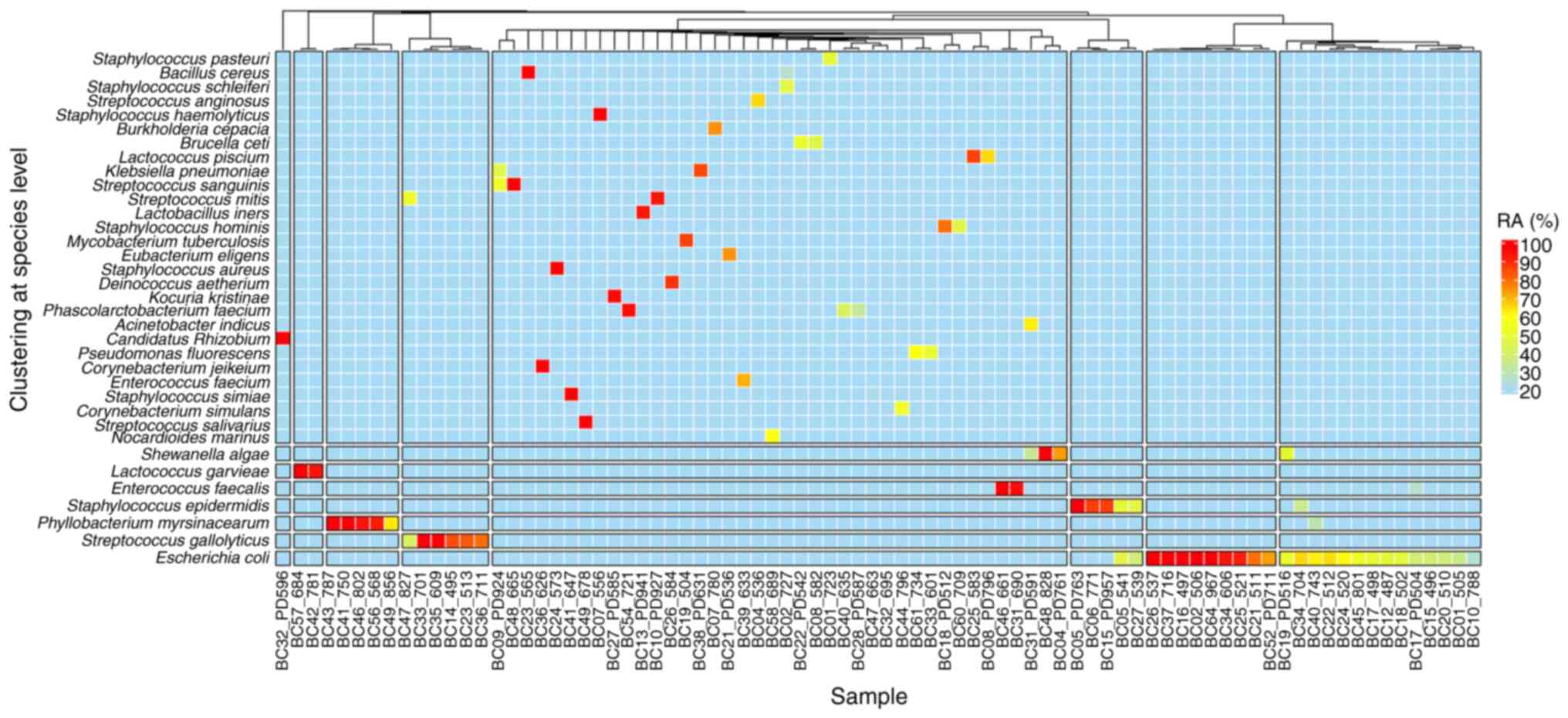
Heatmap analysis was used to visualize the
hierarchical clustering of bacterial diversity, and thereby reveal
the top 35 most abundant bacterial species. All subjects were
divided into eight clusters according to the microbial community
patterns in the samples (Fig. 3).
The dominant bacterial community included Candidatus
Rhizobium (cluster 1), Lactococcus garvieae (cluster 2),
P. myrsinacearum (cluster 3), S. gallolyticus
(cluster 4) and S. epidermidis (cluster 6). E. coli
dominated in clusters 7 and 8, which were distinguished by relative
abundances of >70 and <70%, respectively. The other microbial
community patterns were classified in cluster 5. This result showed
that the overwhelming presence of E. coli in clusters 7 and
8, with obvious differences in relative abundance, could imply a
differential impact on the health of the patient, potentially
associated with varying infection risks or outcomes. This
distinction between the high-abundance (>70%) and
moderate-abundance (<70%) groups of E. coli may point to
different colonization or infection dynamics, suggesting that
cluster 7 could be at a higher risk of infection-related
complications compared with cluster 8(23). The classification of other microbial
community patterns into cluster 5 might represent a mixed or
transitional flora, possibly including organisms that are less
common or less dominant but could still play a role in the catheter
ecosystem, either as commensals or opportunistic pathogens
(24). This diversity within
cluster 5 might also indicate a fluctuating microbial environment
influenced by external factors such as antibiotic use, patient
hygiene, or the procedure of catheter insertion and
maintenance.
Comparison between metagenomic
approach and traditional culture for bacterial identification
Among the 89 PDE samples, bacterial species were
identified from only 56 samples (62.92%) based on the traditional
culture method, whereas all samples (100%) could be classified
through the metagenomic approaches (Fig. 4). In a comparison between
metagenomic and traditional culture methods for bacterial
classification in the 56 samples, concordant results from both
techniques were observed in 42/56 samples (75%). Briefly, the
dominant bacterial species were E. coli (eight cases), S.
epidermidis (six cases), K. pneumoniae (three cases),
S. aureus (three cases), E. faecalis (two cases),
P. aeruginosa (two cases), S. mitis (two cases), and
16 other bacterial species (one case each), as summarized in
Table I. On the other hand,
regarding the remaining 14/56 samples (25%), different results were
demonstrated between the metagenomic approaches and traditional
culture methods, as shown in Table
II. Interestingly, the metagenomic approaches could be applied
for bacterial classification in 33/89 samples (37.08%), which were
negative as far as the traditional culture method was concerned.
The metagenomic results are summarized in Table III.
 | Table ISummary of concordant bacterial
species obtained from both methods. |
Table I
Summary of concordant bacterial
species obtained from both methods.
| Bacterial
species | Samples ID | Number of cases
(%) |
|---|
| Acinetobacter
indicus | PD591 | 1/42 (2.38) |
| Burkholderia
cepacia | 780 | 1/42 (2.38) |
| Candidatus
Rhizobium | PD596 | 1/42 (2.38) |
| Citrobacter
freundii | 485 | 1/42 (2.38) |
| Corynebacterium
simulans | 796 | 1/42 (2.38) |
| Corynebacterium
striatum | 568 | 1/42 (2.38) |
| Corynebacterium
striatum | 633 | 1/42 (2.38) |
| Enterococcus
faecium | | |
| Escherichia
coli | 505, 487, 497, 510,
511, 716, 750, PD711 | 8/42 (19.05) |
| Enterobacter
cloacae | 520 | 1/42 (2.38) |
| Enterococcus
faecalis | 690, 661 | 2/42 (4.76) |
| Klebsiella
pneumoniae | 736, PD924,
PD631 | 3/42 (7.15) |
| Lactococcus
garvieae | 684 | 1/42 (2.38) |
| Pseudomonas
aeruginosa | 734, 601 | 2/42 (4.76) |
| Shewanella
algae | 828 | 1/42 (2.38) |
| Staphylococcus
aureus | 573, 647, 663 | 3/42 (7.15) |
| Staphylococcus
epidermidis | 541, 539, 711, 771,
PD763, PD957 | 6/42 (14.29) |
| Staphylococcus
haemolyticus | 556 | 1/42 (2.38) |
| Staphylococcus
hominis | 709 | 1/42 (2.38) |
| Staphylococcus
pasteuri | 723 | 1/42 (2.38) |
| Staphylococcus
schleiferi | 727 | 1/42 (2.38) |
| Streptococcus
anginosus | 536 | 1/42 (2.38) |
| Streptococcus
gallolyticus | 495 | 1/42 (2.38) |
| Streptococcus
mitis | 827, PD927 | 2/42 (4.76) |
| Table IISummary of different bacterial
species between the metagenomic approach and traditional culture
method. |
Table II
Summary of different bacterial
species between the metagenomic approach and traditional culture
method.
| Sample ID | Traditional culture
method result | Top three of
metagenomic approach result (abundance, %) |
|---|
| 496 | Kocuria
kristinae | Escherichia
coli (38.54%), Ruminococcus gnavus (36.54%),
Prevotella copri (11.45%) |
| 502 | Ochrobactrum
anthropic | Escherichia
coli (48.22%), Methylobacterium dankookense (26.55%),
Clostridium saccharolyticum (7.73%) |
| 506 | Pseudomonas
aeruginosa | Escherichia
coli (98.70%), Butyricicoccus pullicaecorum (0.68%),
Schlesneria paludicola (0.63%) |
| 525 | Staphylococcus
aureus | Catabacter
hongkongensis (28.33%), Saccharofermentans acetigenes
(21.85%), Coprococcus comes (18.24%) |
| 554 |
Corynebacterium sp. | Zhizhongheella
caldifontis (40.20%), Sarcina ventriculi (37.47%),
Achromobacter ruhlandii (5.99%) |
| 582 | Pseudomonas
aeruginosa | Brucella
ceti (48.96%), Oscillibacter valericigenes (21.84%),
Achromobacter xylosoxidans (7.65%) |
| 593 | Rhizobium
radiobacter | Lactobacillus
reuteri (33.81%), Catabacter hongkongensis (21.98%),
Cellulosilyticum lentocellum (17.36%) |
| 665 | Staphylococcus
epidermidis | Streptococcus
sanguinis (100%) |
| 695 | Mycobacterium
tuberculosis | Brachybacterium
conglomeratum (35.84%), Propionibacterium acnes
(26.55%), Escherichia coli (20.58%) |
| 781 | Coagulase
Negative Staphylococcus | Lactococcus
garvieae (93.95%), Cellulosilyticum lentocellum (2.21%),
Escherichia coli (1.24%) |
| 788 | Shewanella
putrefaciens | Escherichia
coli (24.51%), Propionibacterium acnes (22.27%),
Roseburia faecis (17.22%) |
| PD512 | Klebsiella
pneumonia | Staphylococcus
hominis (79.20%), Escherichia coli (17.44%),
Veillonella atypica (0.71%) |
| PD542 | Mycobacterium
tuberculosis | Brucella
ceti (50.73%), Parabacteroides faecis (47.73),
Roseburia intestinalis (1.48%) |
| PD941 | Staphylococcus
haemolyticus | Lactobacillus
iners (93.77%), Pirellula staleyi (4.15%),
Methylobacterium podarium (1.93%) |
| Table IIISummary of bacterial species through
the only metagenomic method. |
Table III
Summary of bacterial species through
the only metagenomic method.
| Sample ID | Top three of the
metagenomic approach result (abundance, %) |
|---|
| 488 | M.
thermophilus (28.32%), C. clostridioforme (22.94%),
P. copri (12.99%) |
| 498 | E. coli
(49.62%), M. tuberculosis (16.36%), A. chartisolvens
(12.73%) |
| 504 | M.
tuberculosis (86.92%), C. segnis (2.80%), E. coli
(2.49%) |
| 512 | E. coli
(64.79%), E. fergusonii (19.04%), M. tuberculosis
(5.14%) |
| 513 | S.
gallolyticus (83.13%), E. coli (12.31%), B. ceti
(3.57%) |
| 521 | E. coli
(92.49%), F. magna (7.51%) |
| 537 | E. coli
(100%) |
| 565 | B. cereus
(99.62%), P. acnes (0.30%), B. weihenstephanensis
(0.08%) |
| 583 | L. piscium
(86.47%), D. aetherius (9.74%), L. raffinolactis
(3.79%) |
| 584 | D. aetherius
(88.51%), E. faecalis (7.62%), C. oryzae (2.08%) |
| 606 | E. coli
(93.77%), M. podarium (4.13%), M. radiotolerans
(1.40%) |
| 609 | S.
gallolyticus (98.93%), L. piscium (1.07%) |
| 626 | C. jeikeium
(99.50%), M. halophilus (0.27%), P. acnes
(0.23%) |
| 635 | P. faecium
(41.69%), P. buccalis (14.21%), O. valericigenes
(8.52%) |
| 678 | S.
salivarius (97.45%), S. warneri (2.36%), E.
faecalis (0.19%) |
| 689 | N. marinus
(59.48%), S. gordonii (31.69%), P. sediminis
(8.11%) |
| 701 | S.
gallolyticus (99.54%), E coli (0.46%) |
| 704 | E. coli
(65.45%), S. epidermidis (34.55%) |
| 721 | P. faecium
(94.92%), B. luteolum (3.95%), P. stutzeri
(0.75%) |
| 743 | E. coli
(62.44%), P. myrsinacearum (29.22%), C. minuta
(8.34%) |
| 787 | P.
myrsinacearum (99.53%), O. pituitosum (0.47%) |
| 796 | L. piscium
(65.22%), S. parauberis (20.05%), L. raffinolactis
(14.73%) |
| 801 | E. coli
(52.48%), A. commune (26.08%), F. saccharivorans
(10.55%) |
| 802 | P.
myrsinacearum (94.42%), B. vesicularis (3.59%), R.
mucilaginosa (1.99%) |
| 856 | P.
myrsinacearum (63.30%), B. aurantiaca (27.09%), B.
vesicularis (4.72%) |
| 967 | E. coli
(98.17%), P. acnes (1.83%) |
| PD504 | E. coli
(37.35%), E. faecalis (25.86%), P. capillosus
(17.96%) |
| PD516 | E. coli
(51.20%), S. algae (48.80%) |
| PD536 | E. eligens
(74.34%), M. podarium (11.61%), S. epidermidis
(7.05%) |
| PD585 | K. kristinae
(95.64%), G. para-adiacens (1.62%), S. suis
(0.71%) |
| PD587 | P. faecium
(32.69%), E. cloacae (14.38%), E. eligens
(8.00%) |
| PD683 | P. sediminis
(40.62%), T. brevis (12.76%), P. staleyi
(11.20%) |
| PD761 | S. algae
(72.93%), C. cellulans (15.33%), N. kribbensis
(5.45%) |
Discussion
Amplicon sequencing using the Oxford Nanopore
Technologies platform is a powerful strategy for microbial
identification, and has been popularly employed for microbiome
analysis in diverse patient clinical samples (25). This sequencing platform offers a
culture-free method that both provides a cost-effective technique
and is associated with a number of essential benefits regarding
long-read data (26). The
amplification and sequencing of the full-length 16S rDNA gene
(~1,500 bp) can allow bacterial identification up to the species
level with high accuracy and sensitivity (27,28).
However, a favorable-quality DNA sample is initially required to
amplify the full-length gene for long-read sequencing; therefore,
one limitation of this approach is the difficulty of achieving
full-length gene amplification in low-quality DNA samples.
In the present study, the DNA in PDE samples was
degraded for several reasons, including being collected without
nucleic acid preservation (NAP), RNA/DNA Stabilization Buffer,
multiple freeze-thaw events, and being kept at -20˚C for a long
period of time (29). Degraded
samples may have insufficient quantities of DNA to amplify the
full-length 16S rDNA gene. To solve this problem, the sample should
be preserved in NAP buffer (30),
and avoiding multiple freeze-thaw steps would be more appropriate
for full-length amplicon sequencing. Normally, V3-V4 hypervariable
region of 16S rDNA gene (~500 bp) is widely used for bacterial
identification studies which limits the taxonomic diversity,
specified only at the genus level (31). For V1-V4 (~800 bp), target sequence
was longer than V3-V4 hypervariable. Therefore, the partial 16S
rDNA gene (V1-V4 region) should be applied for identifying the
bacterial species in samples without the preservation buffer and
low abundance of bacterial DNA. Another factor that may contribute
towards DNA degradation would be the lysis buffer used in the DNA
extraction process. A cell wall is found in the majority of
different species of bacteria, and this is substantially more rigid
than the plasma membrane of mammalian cells. A mild lysis buffer
can therefore be used to ensure that the plasma membrane is
selectively lysed with no resultant damage to the microorganisms.
However, certain microorganisms are more likely to be destroyed by
using a selective lysis buffer, which leads to an undesirably low
DNA quantity for library preparation and sequencing (32).
In the present study, the Chao1 and Shannon
diversity indexes in PDE were relatively lower than those found in
the previous study that investigated the microbial diversity in
peritoneal tissue samples (33).
This result suggested that the bacterial composition in the PDE
sample might be diluted compared with bacterial community in the
peritoneal tissue sample. However, the PDE sample collection is
non-invasive and more convenient process. The present study
demonstrated that the traditional bacterial culture method provided
positive results in only 56 of the 104 samples (53.8%), whereas the
16S rDNA metagenomic approach was able to identify up to 89 samples
(85.6%). Moreover, the current study showed that the same results
were obtained comparing between the traditional culture method and
16S rDNA sequencing in 42/56 samples (75%). Notably, the bacterial
species were classified for the 33 samples (31.73%) that lacked
traditional culture results through the use of 16S rDNA amplicon
sequencing. Considered altogether, the results obtained from the
present study were comparable with those of a recent study
(16), wherein shotgun metagenomic
analysis was performed to identify pathogens in PDE samples based
on the BGISEQ platforms, as summarized in Table IV.
| Table IVThe comparison of positive rate from
bacterial culture and metagenomic analysis between the present
study and a recent report. |
Table IV
The comparison of positive rate from
bacterial culture and metagenomic analysis between the present
study and a recent report.
| Result | Present study | Ye et al,
2022(16) |
|---|
| Positive rate from
culture method | 56/104
(53.85%) | 18/30 (60%) |
| Positive rate from
metagenomic analysis | 89/104
(85.58%) | 26/30 (86.67%) |
| Positive rate from
both techniques | 56/104
(53.85%) | 15/30 (50%) |
| Negative rate from
both techniques | 0/104 (0%) | 1/30 (3.33%) |
In the present study, the 16S rDNA gene sequencing
results showed that Firmicutes, Proteobacteria and Actinobacteria
were the dominant phyla in patients with ESKD, similar to the
findings of a previously published study (34). In line with the findings of the
current study, previous studies identified microbiomes in the
peritoneal tissue of patients with ESKD who harbored a high
abundance of Firmicutes and Proteobacteria (33). The bacterial genera
Escherichia, Streptococcus and Staphylococcus
were also detected in a recent study (16) that used a traditional culture method
and shotgun metagenomic analysis. Therefore, the dominance of E.
coli, P. myrsinacearum, S. gallolyticus, S.
epidermidis and S. algae in the PDE samples in the
present study may be of clinical importance. Moreover, a different
study (35) revealed the causative
microorganisms in PDE samples based on traditional culture that
found both gram-positive bacteria (namely, Staphylococcus,
Streptococcus and Enterococcus) and gram-negative
bacteria (namely, Escherichia, Klebsiella and
Pseudomonas). This finding was consistent in part with those
of the present study.
In general, E. coli is a frequent
gram-negative peritonitis bacterium that can produce the
extended-spectrum β-lactamase that is associated with a poorer
prognosis (36). Interestingly,
PD-associated peritonitis caused by Streptococcus sp. has
been reported from the entry routes into the peritoneal cavity,
including contamination during the exchange or catheter-associated
processes and bacterial translocation (37). A number of studies have shown that
the most common pathogens are coagulase-negative
Staphylococcus species, including S. epidermidis and
S. aureus, which commonly colonize human skin and hands and
may also lead to peritonitis via exit-site and tunnel infections
(38,39). Shewanella sp. are hydrogen
sulfide-producing, motile gram-negative bacilli. The clinical
syndromes that are commonly encountered are skin and soft tissue
infections, including peritoneal catheter-associated infections
(40). Finally, P.
myrsinacearum is a gram-negative bacterium that can cause
infections in humans. As P. myrsinacearum cannot grow on
standard media culture, the identification of this bacterium based
on a metagenomic approach raised several outstanding questions
about whether it is able to cause severe infection in humans
(41).
In summary, the present study demonstrated the
metagenomic analysis based on the partial gene amplification of 16S
rDNA with Oxford Nanopore Technologies, which is suitable for PDE
samples containing low abundance of DNA. The present metagenomic
approach would be useful for monitoring possible bacterial
infections in patients with CKD with peritoneal dialysis. Moreover,
this method might be attractive and applicable for other specimens
with low amount of DNA such as urine, skin and conjunctival
specimens. Furthermore, it can be applied to select appropriate
medicine to reduce antibiotics resistance prone in long-term.
Supplementary Material
The 1% agarose gel electrophoresis of
the representative PCR products using 16S_27F/16S_806R primers
specific to bacterial 16S rDNA (V1-V4 region) amplification. The
triangle indicates the expected band. ‘+’ and ‘-’ indicate the
positive and negative controls, respectively. bp, base pairs; M,
100 bp ladder marker; S1-S4: Representative peritoneal dialysis
effluent samples.
Rarefaction curve analysis. The X-axis
represents the sequencing depth, whereas the Y-axis represents the
Shannon diversity index. Each color line represents different
samples.
Acknowledgements
The authors would like to thank all members of the
Center of Excellence in Systems Microbiology (CESM), Faculty of
Medicine, Chulalongkorn University for technical support and
assistance with the experiments in the present study.
Funding
Funding: The present study was supported by Ratchadapisek Sompot
Fund (grant nos. GA66/034, HEA663000115, HEA663000116 and
RA-MF-26/66), Faculty of Medicine, Chulalongkorn University. The
authors also received a grant from the National Research Council of
Thailand (NRCT) (grant no. N41A660174), Thailand Science Research
and Innovation Fund, Chulalongkorn University (grant no.
CU_FRB65_hea (19) 026_30_07) and the Royal College of Physicians of
Thailand (grant no. 02/66). The present study was supported in part
by the Chulalongkorn University Graduate Scholarship to Commemorate
the 72nd Anniversary of His Majesty King Bhumibol Adulyadej.
Availability of data and materials
The data generated in the present study may be found
in the National Center for Biotechnology Information (NCBI)
Sequence Read Archive (SRA) under accession numbers SRR26930019 to
SRR26930107. The data generated in the present study may be found
in the NCBI GenBank under accession number PRJNA1044279 or at the
following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1044279.
Authors' contributions
SP and TK conceptualized the study. SV conducted the
experiments, analyzed the data, interpreted the results and
prepared the draft version of the manuscript. PK, VS and PS
contributed to data analysis. PC, TS and PP contributed to the
experiments. SP and TK oversaw, revised the final manuscript and
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was conducted in line with the
Declaration of Helsinki (2013), and the protocol of this study was
approved by the Institutional Review Board of the Faculty of
Medicine, Chulalongkorn University (approval no. 0754/2022; IRB no.
0253/65; Bangkok, Thailand). Informed consent was obtained from all
included patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Akchurin OM: Chronic kidney disease and
dietary measures to improve outcomes. Pediatr Clin North Am.
66:247–267. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
NIDDK: Peritoneal Dialysis. Journal
1950.
|
|
3
|
Zimmerman AM: Peritoneal dialysis:
Increasing global utilization as an option for renal replacement
therapy. J Glob Health. 9(020316)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Akoh JA: Peritoneal dialysis associated
infections: An update on diagnosis and management. World J Nephrol.
1:106–122. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Brook NR, White SA, Waller JR and
Nicholson ML: The surgical management of peritoneal dialysis
catheters. Ann R Coll Surg Engl. 86:190–195. 2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Amato D, de Jesus Ventura M, Miranda G,
Leaños B, Alcántara G, Hurtado ME and Paniagua R: Staphylococcal
peritonitis in continuous ambulatory peritoneal dialysis:
Colonization with identical strains at exit site, nose, and hands.
Am J Kidney Dis. 37:43–48. 2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Deng X, Achari A, Federman S, Yu G,
Somasekar S, Bártolo I, Yagi S, Mbala-Kingebeni P, Kapetshi J,
Ahuka-Mundeke S, et al: Metagenomic sequencing with spiked primer
enrichment for viral diagnostics and genomic surveillance. Nat
Microbiol. 5:443–454. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Simner PJ, Miller S and Carroll KC:
Understanding the promises and hurdles of metagenomic
next-generation sequencing as a diagnostic tool for infectious
diseases. Clin Infect Dis. 66:778–788. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rota PA, Oberste MS, Monroe SS, Nix WA,
Campagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen
MH, et al: Characterization of a novel coronavirus associated with
severe acute respiratory syndrome. Science. 300:1394–1399.
2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Greninger AL and Zerr DM: NGSocomial
infections: High-Resolution views of hospital-acquired infections
through genomic epidemiology. J Pediatric Infect Dis Soc. 10
(Supplement 4):S88–S95. 2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chiu CY and Miller SA: Clinical
metagenomics. Nat Rev Genet. 20:341–355. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chiu CY: Viral pathogen discovery. Curr
Opin Microbiol. 16:468–478. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gu W, Deng X, Lee M, Sucu YD, Arevalo S,
Stryke D, Federman S, Gopez A, Reyes K, Zorn K, et al: Rapid
pathogen detection by metagenomic next-generation sequencing of
infected body fluids. Nat Med. 27:115–124. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Greninger AL, Naccache SN, Federman S, Yu
G, Mbala P, Bres V, Stryke D, Bouquet J, Somasekar S, Linnen JM, et
al: Rapid metagenomic identification of viral pathogens in clinical
samples by real-time nanopore sequencing analysis. Genome Med.
7(99)2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nie S, Zhang Q, Chen R, Lin L, Li Z, Sun
Y, Huang J, Feng Z, Cao X, Ye K, et al: Rapid detection of
pathogens of peritoneal dialysis-related peritonitis, especially in
patients who have taken antibiotics, using metagenomic
next-generation sequencing: A pilot study. Ren Fail.
45(2284229)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ye P, Xie C, Wu C, Yu C, Chen Y, Liang Z,
Chen Y, Chen Q and Kong Y: The application of metagenomic
next-generation sequencing for detection of pathogens from dialysis
effluent in peritoneal dialysis-associated peritonitis. Perit Dial
Int. 42:585–590. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wick RR, Judd LM and Holt KE: Performance
of neural network basecalling tools for Oxford Nanopore sequencing.
Genome Biol. 20(129)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lanfear R, Schalamun M, Kainer D, Wang W
and Schwessinger B: MinIONQC: Fast and simple quality control for
MinION sequencing data. Bioinformatics. 35:523–525. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rodriguez-Perez H, Ciuffreda L and Flores
C: NanoCLUST: A species-level analysis of 16S rRNA nanopore
sequencing data. Bioinformatics. 37:1600–1601. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cole JR, Chai B, Marsh TL, Farris RJ, Wang
Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, et
al: The ribosomal database project (RDP-II): Previewing a new
autoaligner that allows regular updates and the new prokaryotic
taxonomy. Nucleic Acids Res. 31:442–443. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bolyen E, Rideout JR, Dillon MR, Bokulich
NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M,
Asnicar F, et al: Reproducible, interactive, scalable and
extensible microbiome data science using QIIME 2. Nat Biotechnol.
37:852–857. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gu Z, Eils R and Schlesner M: Complex
heatmaps reveal patterns and correlations in multidimensional
genomic data. Bioinformatics. 32:2847–2849. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bradley CW, Flavell H, Raybould L, McCoy
H, Dempster L, Holden E and Garvey MI: Reducing Escherichia coli
bacteraemia associated with catheter-associated urinary tract
infections in the secondary care setting. J Hosp Infect.
98:236–237. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Eggimann P, Sax H and Pittet D:
Catheter-related infections. Microbes Infect. 6:1033–1042.
2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Fu Y, Chen Q, Xiong M, Zhao J, Shen S,
Chen L, Pan Y, Li Z and Li Y: Clinical performance of nanopore
targeted sequencing for diagnosing infectious diseases. Microbiol
Spectr. 10(e0027022)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Midha MK, Wu M and Chiu KP: Long-read
sequencing in deciphering human genetics to a greater depth. Hum
Genet. 138:1201–1215. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Matsuo Y, Komiya S, Yasumizu Y, Yasuoka Y,
Mizushima K, Takagi T, Kryukov K, Fukuda A, Morimoto Y, Naito Y, et
al: Full-length 16S rRNA gene amplicon analysis of human gut
microbiota using MinION™ nanopore sequencing confers species-level
resolution. BMC Microbiol. 21(35)2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
D'Andreano S, Cusco A and Francino O:
Rapid and real-time identification of fungi up to species level
with long amplicon nanopore sequencing from clinical samples. Biol
Methods Protoc. 6(bpaa026)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Monger XC, Saucier L, Gilbert AA and
Vincent AT: Stabilization of swine faecal samples influences
taxonomic and functional results in microbiome analyses. MethodsX.
9(101716)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Menke S, Gillingham MA, Wilhelm K and
Sommer S: Home-Made cost effective preservation buffer is a better
alternative to commercial preservation methods for microbiome
research. Front Microbiol. 8(102)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jeong J, Yun K, Mun S, Chung WH, Choi SY,
Nam YD, Lim MY, Hong CP, Park C, Ahn YJ and Han K: The effect of
taxonomic classification by full-length 16S rRNA sequencing with a
synthetic long-read technology. Sci Rep. 11(1727)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shi Y, Wang G, Lau HC and Yu J:
Metagenomic sequencing for microbial DNA in human samples: Emerging
technological advances. Int J Mol Sci. 23(2181)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Simoes-Silva L, Araujo R, Pestana M,
Soares-Silva I and Sampaio-Maia B: Peritoneal microbiome in
end-stage renal disease patients and the impact of peritoneal
dialysis therapy. Microorganisms. 8(173)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan
J, DeSantis TZ, Ni Z, Nguyen TH and Andersen GL: Chronic kidney
disease alters intestinal microbial flora. Kidney Int. 83:308–315.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Dzekova-Vidimliski P, Nikolov IG,
Gjorgjievski N, Selim G, Trajceska L, Stojanoska A,
Rambabova-Bushljetik I, Simeonov R and Stojkovski L: Peritoneal
dialysis-related peritonitis: Rate, clinical outcomes and patient
survival. Pril (Makedon Akad Nauk Umet Odd Med Nauki). 42:47–55.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Feng X, Yang X, Yi C, Guo Q, Mao H, Jiang
Z, Li Z, Chen D, Cui Y and Yu X: Escherichia coli Peritonitis in
peritoneal dialysis: The prevalence, antibiotic resistance and
clinical outcomes in a South China dialysis center. Perit Dial Int.
34:308–316. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chao CT, Lee SY, Yang WS, Chen HW, Fang
CC, Yen CJ, Chiang CK, Hung KY and Huang JW: Viridans streptococci
in peritoneal dialysis peritonitis: Clinical courses and long-term
outcomes. Perit Dial Int. 35:333–341. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Salzer WL: Peritoneal dialysis-related
peritonitis: Challenges and solutions. Int J Nephrol Renovasc Dis.
11:173–186. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Gadola L, Poggi C, Dominguez P, Poggio MV,
Lungo E and Cardozo C: Risk factors and prevention of peritoneal
dialysis-related peritonitis. Perit Dial Int. 39:119–125.
2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yan Y, Chai X, Chen Y and Zhang X: The
fulminating course of infection caused by shewanella algae: A case
report. Infect Drug Resist. 15:1645–1650. 2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hughes G, Pallard C, Chavdab S and Davida
MD: The first case report of human infection with Phyllobacterium
myrsinacearum causing spondylodiscitis. Clin Infec Pract.
7-8(100029)2020.
|