Introduction
Uric acid is the end product of purine metabolism in
humans and higher primates, which is mainly synthesized in the
liver and is predominantly excreted by the kidneys. Under normal
physiological conditions, serum uric acid concentrations range from
3.5 to 7.2 mg/dl in men, and from 2.6 to 6.0 mg/l in women; these
levels are maintained through a tightly regulated balance between
uric acid production and excretion (1). When this balance is disrupted, leading
to elevated levels of uric acid in the blood, hyperuricemia occurs
(2,3). Clinically, hyperuricemia is defined as
serum uric acid concentrations ≥7 mg/dl (0.42 mmol/l) at
physiological temperature and pH (4).
Chronic hyperuricemia is implicated in the
development of various pathophysiological conditions, including
hypertension, chronic kidney disease, cardiovascular disease,
nonalcoholic fatty liver disease, metabolic syndrome and diabetes
(5,6). The association between hyperuricemia
and these conditions is well known, making it a significant risk
factor for cardiovascular and renal diseases. Despite this, the
precise serum uric acid concentration at which the risk begins to
increase remains unclear. Emerging evidence has suggested that the
threshold for increased risk may be lower than previously
considered, potentially <6 mg/dl (5). Consequently, this issue continues to
be a focal point for clinical research, as understanding the exact
uric acid levels that pose a risk is crucial for developing
effective treatment and prevention strategies (5-7).
Uric acid can interact with reactive oxygen species (ROS), such as
hydroxyl radicals and hypochlorous acid, transforming them into
less harmful substances such as allantoin, allantoate, glyoxylate,
urea and oxalate (8). This
antioxidant capability has been demonstrated in various
experimental studies, both in vitro and in vivo,
including studies on isolated organs and the human lung (9). Moreover, uric acid serves as an
oxidizable co-substrate for cyclooxygenase enzymes, thereby
contributing to the reduction of oxidative stress and the
maintenance of endothelial function, particularly in coronary
circulation (10). Despite its
beneficial antioxidant properties, elevated uric acid levels, or
hyperuricemia, are linked to numerous adverse health
conditions.
Epidemiological studies have consistently identified
hyperuricemia as a key contributor to hypertension (11-14).
Experimental research in a rat model has shown that dietary
supplementation with oxonic acid can lead to increased plasma uric
acid levels and subsequent increased blood pressure (15-17).
Additionally, hyperuricemia has been associated with endothelial
dysfunction, impaired vasodilation and vascular injury, which are
precursors to cardiovascular diseases, including hypertension and
atherosclerosis (18-21).
These findings highlight the complex role of uric acid in vascular
biology, where its antioxidant functions are unclear due to its
potential to contribute to vascular damage under hyperuricemic
conditions. The exacerbation of endothelial dysfunction and
vascular injury in the presence of elevated uric acid levels
suggests a critical need to understand the underlying molecular
mechanisms. Further research is needed to identify targeted
therapeutic approaches that potentially reduce the deleterious
effects of hyperuricemia.
Vascular smooth muscle cells (VSMCs) and endothelial
cells (ECs), are integral components of blood vessels, serving
crucial roles in vasoconstriction, vascular tone regulation and the
development of vasculature (22).
Nitric oxide (NO), a key cellular signaling molecule involved in
both physiological and pathological processes, mediates
vasodilation. Under normal conditions, NO is synthesized by
endothelial NO synthase in ECs, diffuses to adjacent VSMCs and
induces vasodilation (22-24).
However, under oxidative stress, NO reacts with superoxide anions
to form peroxynitrite (25,26). Additionally, uric acid leads to a
reduction in NO levels because of the formation of peroxynitrite in
vascular cells (27,28). Peroxynitrite is a potent oxidant
known to cause oxidative damage and nitrosative stress to
macromolecules, such as proteins, lipids and DNA (25,29,30).
Previous studies have demonstrated that elevated uric acid levels
decrease NO production in human umbilical vein ECs (HUVECs)
(31-34).
Furthermore, it has previously been indicated that uric acid
increases oxidative stress, which triggers a phenotypic transition
in vascular ECs (21). While the
deleterious effects of uric acid-induced NO depletion and increased
oxidative stress on EC function have been demonstrated (20,35-37),
emerging evidence has suggested that high uric acid conditions may
also affect VSMCs (38,39). However, the specific impact of uric
acid on NO levels in VSMCs remains elusive. Further studies are
required to elucidate the mechanisms through which uric acid
influences NO bioavailability in VSMCs, and to understand the
implications for vascular health and disease.
The p53 gene is widely recognized as a tumor
suppressor gene, playing a pivotal role in regulating key cellular
processes, including cell cycle control, DNA repair, cell
proliferation, apoptosis, aging and oxidative stress response,
which are associated with diseases such as diabetes, cancer and
hypertension (40,41). In VSMCs, p53 has been shown to
promote senescence and apoptosis, and is actively involved in the
pathogenesis of atherosclerotic plaques (42). However, there are conflicting
reports in the literature regarding the role of p53 in VSMCs.
Recent data have revealed that p53 deficiency reduces VSMCs
senescence and calcification, and knockdown of p53 can decrease
mitochondrial ROS (43), suggesting
that it protects VSMCs against oxidative stress. Moreover, p53 has
been shown to protect VSMCs from NO-mediated oxidative stress
(44). Previous findings have also
revealed that p53 can be directly targeted by uric acid (45). To the best of our knowledge,
although numerous aspects of uric acid-induced cell proliferation
(38,39,46),
inflammation (47,48) and oxidative stress (38,48)
have been studied in VSMCs, the effect of uric acid on p53 has not
yet been demonstrated. The interplay between uric acid and p53 in
VSMCs is of particular interest, given the potential implications
for understanding how uric acid influences cellular senescence,
oxidative stress and vascular pathophysiology.
In the present study, it was hypothesized that uric
acid could elevate oxidative stress in rat VSMCs in a dose- and
time-dependent manner. To investigate this hypothesis, the effects
of various concentrations of uric acid on oxidative stress markers,
including protein carbonylation, thiobarbituric acid reactive
substances (TBARs) and superoxide anion levels, were examined.
Additionally, the protein expression levels of p53 and NO levels
were assessed in rat VSMCs. By comparing these parameters, the
present study aimed to understand the association between uric acid
exposure and oxidative stress, as well as its impact on p53
expression and NO levels over time and across different doses. The
present findings provide preliminary novel insights into oxidative
stress responses induced by uric acid and highlight the potential
role of uric acid in mediating vascular cell function.
Materials and methods
Reagents and antibodies
Uric acid, fetal bovine serum (FBS),
penicillin-streptomycin, HEPES, elastase, collagenase, cytochrome
c, superoxide dismutase (SOD) and bovine serum albumin (BSA)
were purchased from Sigma-Aldrich (Merck KGaA). DMEM (cat. no.
E0500-100) was obtained from Cegrogen Biotech GmbH and Hank's
balanced salt solution (HBSS) was obtained from Biochrom, Ltd.
Nitrate/Nitrite (NO detection) Colorimetric Assay Kit (cat. no.
780001), TBARS Assay Kit (cat. no. 10009055) and Protein Carbonyl
Assay Kit (cat. no. 10005020) were purchased from Cayman Chemical
Company. Primary antibodies against p53 (1C12) (cat. no. 2524) and
β-actin (cat. no. 4967), as well as secondary antibodies
[horseradish peroxidase (HRP)-linked goat anti-rabbit
immunoglobulin G (IgG) (cat. no. 7074) and anti-mouse IgG (cat. no.
7076)] were purchased from Cell Signaling Technology, Inc.
Isolation and culture of primary rat
vascular smooth muscle cells
All animal experiments were performed according to
the Guide for the Care and Use of Laboratory Animals (49) following experimental protocols
approved by the Local Committee on Animal Research Ethics at
Akdeniz University (approval no. 727/2018.01.024; Antalya, Turkey).
For the present study, a total of 4 male Wistar rats (age, 8-10
weeks; weight, 200-300 g) were used. The male Wistar rats were
obtained from the Local Committee on Animal Research Ethics at
Akdeniz University. The rats were housed in a controlled
environment with a temperature of 22±2˚C, a relative humidity of
50±10% and a 12-h light/dark cycle. The rats had ad libitum
access to standard chow and water. According to the latest
guidelines from the American Veterinary Medical Association
(50) and guidelines adopted by
institutions such as Boston University (51) and the University of Maryland
(52), the rats were anesthetized
by intraperitoneal injection of ketamine (80 mg/kg) and xylazine
(10 mg/kg), in compliance with approved ethical standards (53,54).
Upon confirmation of deep anesthesia, which was verified by the
lack of response to painful stimuli and absence of the corneal
reflex, the chest cavity was exposed to allow access to the aorta.
The aorta was then carefully dissected, the adventitial layer was
meticulously removed using forceps, and the tissue was subsequently
transferred to cell culture dishes under sterile conditions. VSMCs
were isolated from the dissected aorta using enzymatic dissociation
solution [HEPES dissolved in HBSS (15 mM, pH: 7.2-7.3), BSA (2
mg/ml), CaCl2 (0.2 mM), Soybean Trypsin Inhibitor (0.25
mg/ml), elastase (0.0625 mg/ml) and collagenase (0.5 mg/ml)]
(39). After the isolation of
VSMCs, while the rats were still under deep anesthesia, they were
sacrificed by cervical dislocation to ensure a humane and painless
death. The isolated cells were subsequently transferred to cell
culture dishes and cultured as previously described (39,47,55).
VSMCs were maintained in DMEM supplemented with 10% FBS and 1%
penicillin-streptomycin, and were cultured at 37˚C and 5%
CO2. VSMCs in passages 3 to 5 were used in all
experiments and the cells were incubated up to 70-80% density.
After the control and experimental groups were formed, all cells
were incubated with FBS-free DMEM overnight for serum starvation.
Uric acid was prepared by filtration through 0.2-mm sterile filters
and VSMCs were stimulated at 37˚C with different uric acid doses
(0-50 mg/dl) for various durations (1-24 h), excluding the control
groups.
NO determination
Once cells reached 80% confluence, NO levels were
detected. NO (total nitrate + nitrite) levels were determined using
a colorimetric assay kit that included Griess reagents (Griess 1
and 2), according to the manufacturer's instructions. NO content
was determined as the total value measured in the presence of cells
minus the value determined from the media alone in the absence of
cell growth, according to the manufacturer's protocol. Each sample
absorbance was measured by spectrophotometry (540 nm) and NO
concentrations were calculated using a standard curve. The levels
of NO are shown in µM. Experiments were repeated four times and the
results are presented as the mean ± standard error of the mean
(SEM).
Superoxide anion accumulation
levels
For the determination of superoxide anion
production, after reaching 80% cell confluence, the Görlach method
of spectrophotometric SOD-inhibitable reduction of cytochrome
c was performed for each sample, with a blank for each one
(negative control), as previously described (47,56).
VSMCs were cultured in 12-well plates for 48 h. Superoxide anion
related to cytochrome c reduction was calculated for each
sample by measuring between cells incubated with SOD and without
SOD. Superoxide anion accumulation levels are shown in nmol/µg.
Experiments were repeated five times and the results are presented
as the mean ± SEM.
Quantification of TBARS levels
Lipid peroxidation was detected by measuring the
amount of malondialdehyde (MDA)-TBA adduct in the cell homogenates.
The cells were collected (2x107) with 1 ml PBS buffer
according to the whole cell lysis procedure indicated in the TBARS
assay kit. The cells were homogenized on ice with an ultrasonic
homogenizer (UW2070; BANDELIN electronic GmbH & Co. KG), SDS
was then added and the cells were mixed with the color reagent (TBA
in acetic acid and sodium hydroxide) in a boiling water bath for 1
h, cooled in an ice-water bath, and then incubated on ice. After
cooling, the sample was centrifuged at 1,600 x g for 10 min at 4˚C
and was maintained at room temperature for 30 min. TBARS was
measured by spectrophotometry (530 nm) and each sample
concentration was determined using the MDA colorimetric standard
curve. Results are expressed as µM. Experiments were repeated three
times and the results are presented as the mean ± SEM.
Quantification of protein carbonyl
content
The concentration of protein carbonyl was determined
spectrophotometrically using a Protein Carbonyl Assay Kit according
to the manufacturer's instructions. Briefly, cells at 80%
confluence were collected and then homogenized in phosphate buffer
(pH 6.7; containing 1 mM EDTA), after which, the sample was
centrifuged at 10,000 x g for 15 min at 4˚C and the supernatant was
removed. The lysates were incubated with dinitrophenylhydrazine for
1 h at room temperature in the dark. The protein was precipitated
twice with trichloroacetic acid (first 20%, second 10%) and was
then washed in an ethanol/ethyl acetate mixture. After being
washed, the sample was resuspended in guanidine hydrochloride and
centrifuged at 10,000 x g for 10 min at 4˚C. The protein carbonyl
content was measured at 360 nm using spectrophotometry and was
calculated according to the manufacturer's instructions. Results
are expressed as nmol/mg protein. The protein content was
determined using the BCA Protein Assay Kit (Takara Bio, Inc.), with
BSA solution as the standard. This assay had a detection limit of
1-10 mg protein. Experiments were repeated three times and the
results are presented the mean ± SEM.
Western blot analysis
Primary rat VSMCs were seeded in 6-well plates.
After the cells reached 80% confluence, sample preparation for
western blotting was performed using lysis buffer [50 mmol/l HEPES,
50 mmol/l NaCl, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 1.5
mmol/l MgCl2, 1 mmol/l EDTA, 10 mmol/l sodium pyrophosphate, 1
mmol/l Na3VO4, 100 mmol/l NaF, 30 mmol/l
2-(p-nitrophenyl) phosphate, 1 mmol/l phenylmethylsulfonyl
fluoride, 10 mg/ml leupeptin and 10 mg/ml aprotinin (pH 7.4)] as
previously described (39). After
the sample protein concentrations were calculated using the BCA
Protein Assay Kit, total proteins (25 µg) were separated by
SDS-PAGE on 10% gels and were then transferred onto 0.2-µm
nitrocellulose membranes (Whatman plc; Cytiva). The membranes were
blocked for 1 h at room temperature with 5% w/v nonfat dry milk
solution, followed by incubation with primary antibodies against
p53 (1:1,000) and β-actin (1:5,000) at 4˚C overnight. The following
day, membranes were washed three times for 5 min in Tris-buffered
saline with 0.01% Tween-20 (TBST) and then incubated with
anti-mouse and anti-rabbit secondary antibodies for 2 h at room
temperature. Subsequently, the membranes were washed three times
for 5 min in TBST. The protein bands were detected using an
enhanced chemiluminescence reagent-based Super Signal West Pico HRP
Substrate System (Thermo Fisher Scientific, Inc.).
Semi-quantification of the protein bands was performed using Alpha
Digi Doc 1,000 Gel Documentation Unit (AlphaEaseFC™; Alpha Innotech
Corporation). After stripping, the membranes were probed with
anti-β-actin antibody for the same duration and at the same
temperature as with the anti-p53 antibody to confirm equal protein
loading. All experiments were performed in triplicate.
Statistical analysis
Statistical analysis was performed with GraphPad
Prism (version 8.01; Dotmatics). The results are presented as the
mean ± SEM. The differences among the control and experimental
groups were evaluated by one-way ANOVA followed by Dunnett's
multiple comparisons test. P<0.05 was considered to indicate a
statistically significant difference.
Results
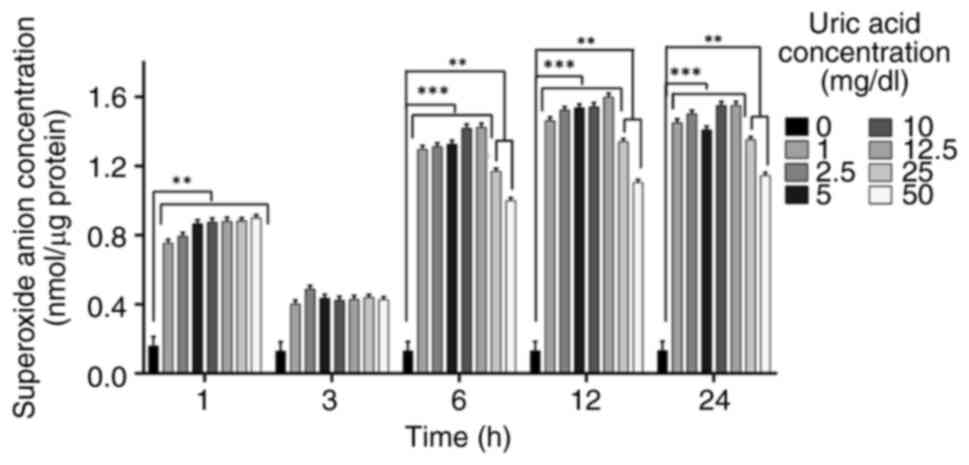
Superoxide anion accumulation is
affected by uric acid in VSMCs independent of dose
Uric acid stimulation can promote the occurrence of
oxidative stress in VSMCs and superoxide anion release is an
upstream target mediator of oxidative stress (57-59).
It was thus hypothesized that uric acid may promote the occurrence
of superoxide anion release by upregulating oxidative stress. To
understand whether superoxide anion release is affected by uric
acid in VSMCs in a dose- and time-dependent manner, the
accumulation of superoxide anion was determined using the Görlach
method (56). Uric acid increased
superoxide anion release at 1, 6, 12 and 24 h in a dose-independent
manner compared with that in the control groups, although there was
no significant difference observed at 3 h (Fig. 1). All uric acid doses decreased
superoxide anion levels at 1, 3, 6, 12, and 24 h compared with the
control; however, the increase observed at 3 h was less pronounced
than at the other time points. In addition, in response to 25 and
50 mg/dl uric acid, superoxide anion accumulation was decreased
compared with in response to the other uric acid concentrations (1,
2.5, 5, 10 and 12.5 mg/dl uric acid) at 6, 12 and 24 h.
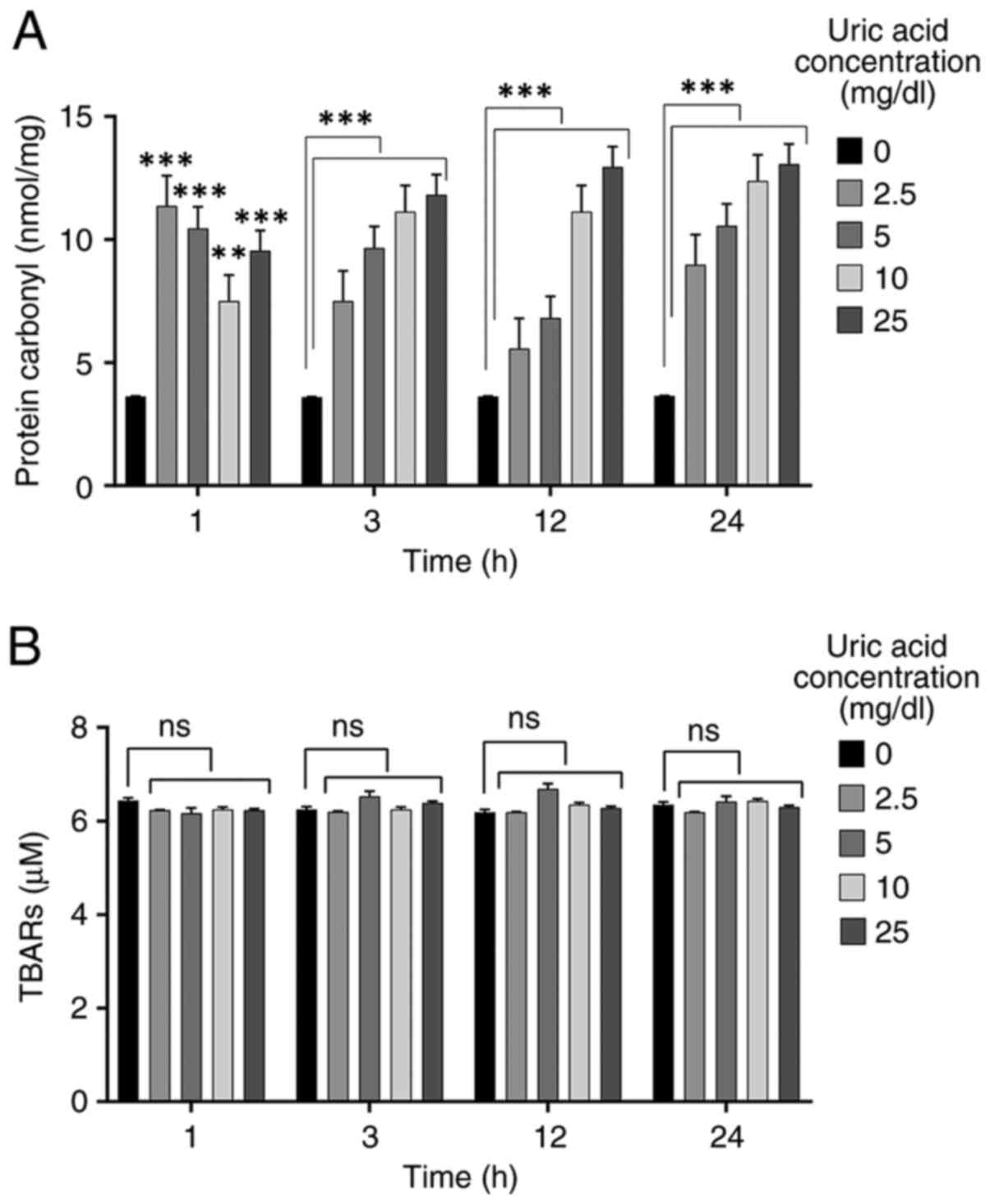
Uric acid promotes protein
carbonylation but does not affect lipid peroxidation
High levels of uric acid can be a key regulator for
oxidative stress (21,35). In order to evaluate the effects of
uric acid on oxidative stress in VSMCs, protein carbonylation and
lipid peroxidation assays were performed. VSMCs were incubated with
different uric acid concentrations (2.5, 5, 10 and 25 mg/dl) for
various durations (1, 3, 12 and 24 h). The control group (0 mg/dl
uric acid concentration) was not treated with uric acid. Treatment
with all uric acid concentrations caused a significant increase in
protein carbonyl levels at 1 h compared with those in the control
group, but 10 mg/dl uric acid dose resulted in a reduction compared
with the other doses (2.5, 5 and 25 mg/dl uric acid) (Fig. 2A). Protein carbonylation in VSMCs
was significantly increased by all concentrations of uric acid at
3, 12 and 24 h compared with that in the control groups in a
dose-dependent manner. As shown in Fig.
2B, uric acid stimulation had no effect on TBARS levels in
VSMCs compared with those in the control group, thus indicating
that no dose of uric acid promoted lipid peroxidation in VSMCs.
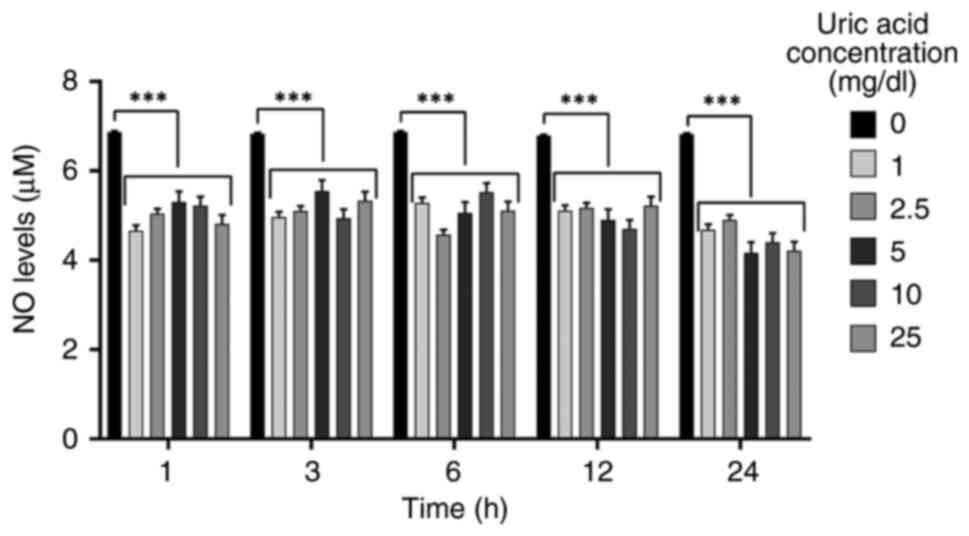
Uric acid decreases NO levels
independent of dose and time
NO is a vasodilator that modulates important
processes, such as vascular tone, inflammation and
oxidation-sensitive mechanisms (60,61).
To determine if higher uric acid concentrations modify NO levels in
VSMCs, NO levels were analyzed in response to different uric acid
doses for various durations. As shown in Fig. 3, all uric acid doses significantly
diminished NO levels in a dose- and time-independent manner.
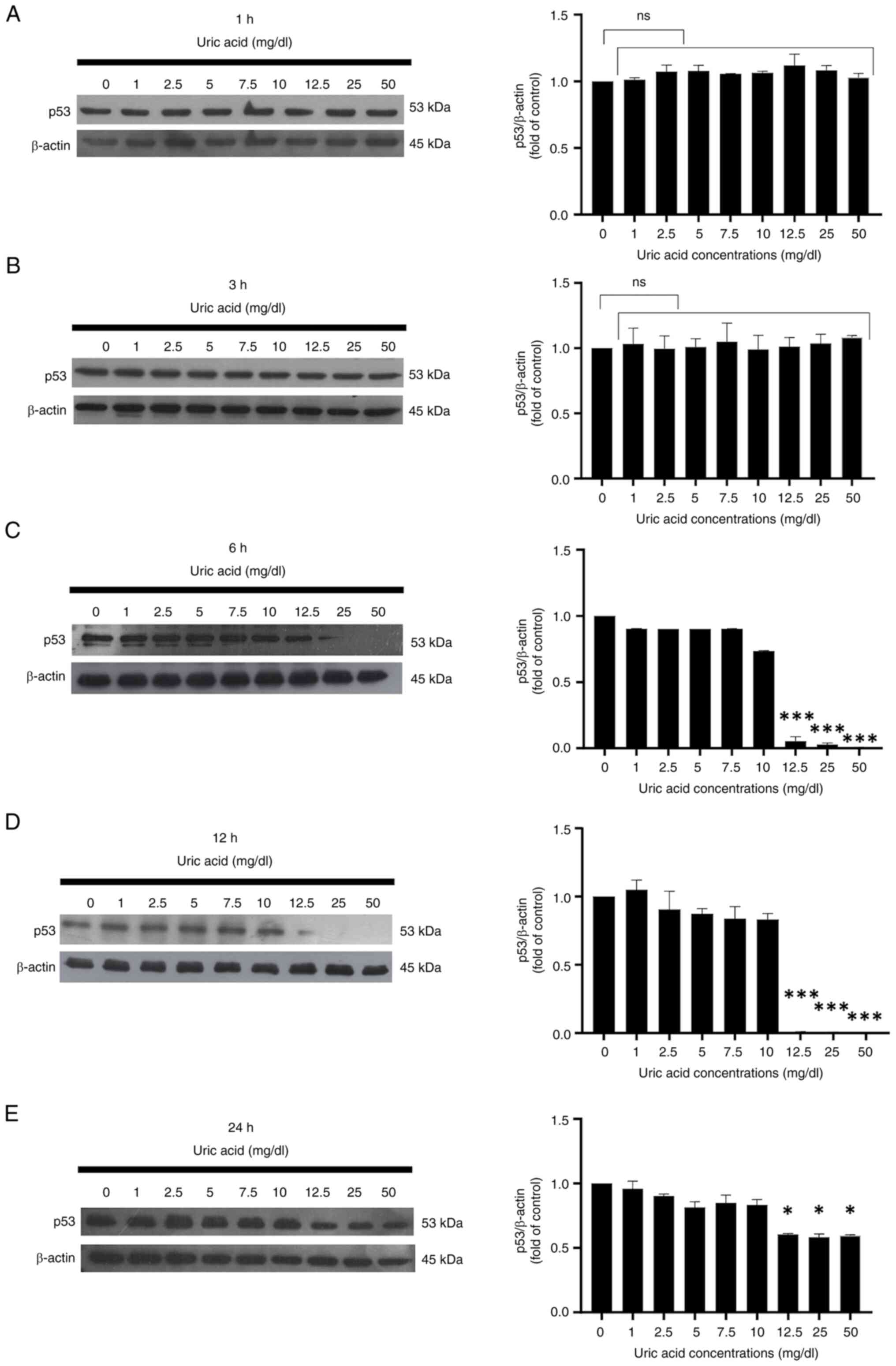
Long-term exposure to high uric acid
levels suppresses p53 expression in VSMCs
Uric acid affected the protein expression levels of
p53 in rat primary VSMCs in a time-dependent manner. As determined
by western blotting, it was determined that all uric acid
concentrations did not affect the expression levels of p53 in VSMCs
at 1 and 3 h compared with those in the control group (0 mg/dl uric
acid) (Fig. 4A and B). As shown in Fig. 4C, p53 expression was suppressed in
VSMCs stimulated with high uric acid doses (12.5, 25 and 50 mg/dl
uric acid concentrations) at 6 h. Long-term uric acid stimulation
of VSMCs indicated that high concentrations of uric acid (12.5, 25
and 50 mg/dl uric acid) abolished p53 expression at 12 h (Fig. 4D), but the same doses of uric acid
only slightly reduced the protein expression levels of p53 at 24 h
(Fig. 4E).
Discussion
Uric acid is known to induce oxidative stress, which
can pathologically contribute to hypertension in VSMCs (62). Given its significant role in causing
hypertension, the vascular damage and endothelial injury induced by
high uric acid levels was further assessed in the present study.
VSMCs have a crucial role in maintaining endothelial homeostasis
and in the development of blood vessels (22). Although research on the effects of
uric acid on VSMCs is insufficient, numerous in vitro and
in vivo studies have demonstrated that ECs continuously
interact with VSMCs. These studies also showed that large amounts
of NO produced by ECs diffuse into VSMCs, where NO is essential for
regulating vascular contraction and relaxation (63-65).
Despite these findings, the precise molecular mechanisms by which
NO operates in uric acid-stimulated VSMCs under oxidative stress
conditions remain elusive. Understanding these mechanisms is
crucial as NO serves a pivotal role in vascular function and
integrity. The interactions between ECs and VSMCs, particularly the
transfer and effects of NO, are central to vascular health, and
disruptions can lead to pathological states. Further research is
needed to elucidate the pathways involved in NO signaling within
VSMCs exposed to high levels of uric acid, as this knowledge could
contribute to the development of therapeutic strategies aimed at
mitigating uric acid-induced vascular damage and hypertension.
Oxidative stress is a pathological condition
characterized by the excessive production of ROS, such as
superoxide anion, hydroxyl radicals and hydrogen peroxide, along
with oxidative damage to macromolecules (66-69).
This process has been identified as a significant clinical risk
factor in contributing to vascular damage, endothelial injury and
the progression of vascular diseases (70-73).
Emerging evidence has consistently demonstrated that uric acid is a
critical factor in the development of endothelial dysfunction by
regulating the oxidative stress of HUVECs (18,21,35,36).
Specifically, studies have shown that uric acid stimulation leads
to increased oxidative stress. Research has also indicated that
uric acid-stimulated VSMCs have a marked increase in ROS
production, and uric acid has been shown to affect oxidative
stress-related signaling pathways within VSMCs (48,74).
The present study focused on the effect of uric acid
on various oxidative stress parameters, including protein
carbonylation, lipid peroxidation and superoxide anion levels in
VSMCs after 1-24 h of treatment. Uric acid increased protein
carbonylation levels in a dose-dependent manner. Specifically,
protein carbonyl levels showed a statistically significant increase
even at the 1-h time point, and this increase was almost maintained
at 3, 6, 12 and 24 h, especially in response to higher
concentrations of uric acid. The results at the short-term time
point (1 h) suggested that uric acid may induce oxidative stress,
leading to protein carbonylation in VSMCs and a high reactivity of
uric acid with cellular proteins indicating that it may rapidly
cause oxidative modifications. Additionally, the results in
response to prolonged exposure (3, 12 and 24 h) provide a more
comprehensive understanding of the impact of uric acid on oxidative
stress. These findings emphasize the necessity of examining both
short-term and long-term exposures to fully elucidate the
biochemical pathways and molecular mechanisms. By contrast, the
present analysis of lipid peroxidation, as measured by TBARs
levels, revealed that uric acid did not significantly affect lipid
peroxidation in VSMCs. This indicated that uric acid may
preferentially induce oxidative stress in proteins rather than
lipids. This selectivity may be attributed to differences in the
susceptibility of proteins and lipids to oxidative damage, or it
may be related to the specific localization of uric acid within
cellular compartments. Moreover, VSMCs may possess robust
antioxidant defense mechanisms, such as glutathione peroxidase and
catalase, which effectively mitigate lipid peroxidation but are
less effective against protein oxidation. This could explain the
observed increase in protein carbonylation despite unchanged TBARs
levels (75). To the best of our
knowledge, the present study is the first to present data on the
effects of uric acid stimulation on protein carbonylation and lipid
peroxidation in rat primary VSMCs.
The present study indicated that all doses of uric
acid increased superoxide anion release compared with that in the
control groups at most time points (1-24 h, with the exception of
at 3 h). Previous studies showed that a uric acid dose of 5 mg/dl
significantly increased superoxide anion accumulation at 1 h in
primary rat VSMCs (47). Consistent
with previous findings (47), the
current results demonstrated that all doses of uric acid caused a
transient reduction of superoxide anion production in VSMCs at 3 h
compared with at the other time points (1, 6, 12 and 24 h).
Although there is no direct evidence to explain the transient
decrease in superoxide anion levels at 3 h, it may be that the
protective effect of the cells against oxidative damage is related
to its ability to reduce oxidative stress in the early time
intervals. These findings have the potential to provide evidence
for the time-dependent effects of uric acid on superoxide anion
production in VSMCs.
p53 is a central mediator of oxidative stress and
apoptosis signaling in vascular functions (76), but its role in the pathogenesis of
vascular damage remains insufficiently understood. Previous studies
have linked p53 to apoptosis in VSMCs (77,78),
although these investigations have been largely confined to the
process of atherosclerotic plaque formation. Notably, p53 exhibits
bidirectional functions in various biological processes, and its
paradoxical role in metabolic pathways is attributed to the
context-dependent nature of its activity (79,80).
Despite this complexity, p53 signaling is recognized as a crucial
regulator of oxidative stress, proliferation and inflammation.
Furthermore, p53 activity has been implicated in uric acid-induced
oxidative stress (45,81,82).
Previous studies have shown that in response to mild ROS
concentrations, p53 promotes cell survival by exerting an
anti-oxidative effect to protect cells from damage. However, when
cells are exposed to excessive and/or prolonged ROS levels, which
can cause uncontrollable damage, p53 activity is inhibited by ROS,
leading to cell death pathways to protect adjacent undamaged cells
(83,84). In light of these findings, the
present study aimed to evaluate the changes in p53 protein
signaling in VSMCs stimulated with uric acid in a dose- and
time-dependent manner. The results demonstrated that p53 protein
expression was significantly suppressed by high doses of uric acid
(12.5, 25 and 50 mg/dl) during prolonged stimulations (6-24 h).
These results provided preliminary findings indicating a dose- and
time-dependent relationship between uric acid exposure and p53
activity in VSMCs.; with p53 expression remaining stable at early
time points (1 and 3 h) but progressively decreasing at 6 and 12 h,
especially in response to higher uric acid concentrations.
Furthermore, when comparing the results of p53 protein expression
with oxidative stress parameters, it was observed that the
suppression of p53 protein expression was associated with increased
superoxide anion accumulation during long-term stimulation of high
uric acid doses. These findings suggested that the accumulation of
superoxide anions induced by uric acid may be associated with the
suppression of p53 activation in VSMCs. Although the data provide
valuable information about the molecular mechanisms by which uric
acid affects vascular cell function, further studies are needed to
explore p53 and oxidative stress pathways in vascular diseases.
It has been determined that NO-mediated apoptosis is
increased by p53 deficiency in VSMCs (85,86).
Furthermore, it has been shown that p53 deficiency can be increased
by NO-mediated oxidative stress (44). These findings highlight the
protective role of p53 in VSMCs; however, the precise relationship
between p53 and NO in these cells remains unclear. It was
hypothesized that p53 might also modulate the levels of oxidative
stress in VSMCs, thereby controlling NO levels, given that one of
the main functions of p53 is the regulation of oxidative stress. To
explore this hypothesis, VSMCs were stimulated with various doses
of uric acid over different time periods and superoxide anion,
TBARS and protein carbonylation levels were measured. The results
of the present study showed that uric acid concentrations decreased
NO levels in VSMCs in a dose-independent manner at all time points.
Based on these findings, it was concluded that the protective
effect of p53 against uric acid-induced oxidative stress in VSMCs
may not be mediated through the regulation of NO levels. This
suggests that p53 may exert its protective effects through
alternative pathways or mechanisms that do not directly involve NO.
Additionally, the study observed increases in protein carbonyl
levels and superoxide anion in response to uric acid exposure,
which are all indicators of oxidative stress. Furthermore, TBARS
was not directly related to the concentration of uric acid,
indicating that TBARS levels were not sensitive to changes in uric
acid concentration. This conclusion is visually summarized in
Fig. 5, which illustrates the
complex interactions between uric acid, oxidative stress markers
and p53 activity.
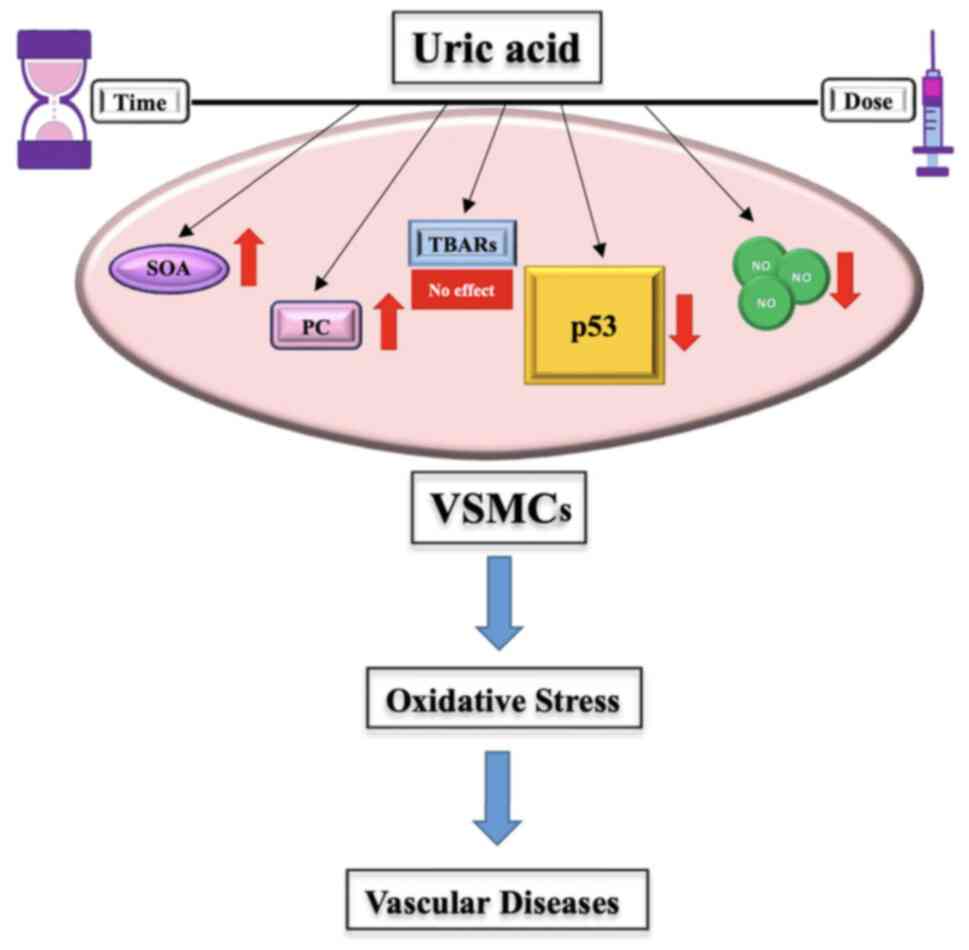 | Figure 5Effects of uric acid on oxidative
stress in VSMCs. Uric acid significantly promoted an increase in
SOA levels and PC, which are key indicators of oxidative stress. It
also resulted in a marked reduction in NO concentrations, a
molecule essential for vascular homeostasis. Additionally, uric
acid did not significantly alter TBARs levels. Uric acid
exacerbated oxidative stress, with its inhibitory effects on p53
potentially contributing to this process. Given these observations,
uric acid may serve as a reliable biomarker for oxidative stress in
VSMCs. VSMCs, vascular smooth muscle cells; TBARs, thiobarbituric
acid reactive substances; SOA, superoxide anion; PC, protein
carbonylation; NO, nitric oxide. |
To the best of our knowledge, the present study is
the first to show that uric acid affected NO levels in VSMCs in a
dose- and time-independent manner, while also providing
comprehensive evidence that uric acid increased superoxide anion
levels at 1, 3, 6, 12 and 24 h, with a less pronounced increase at
3 h compared with the other time points, and its dose-dependent
effects on protein carbonyl levels. The results demonstrated that
the doses of uric acid that induced accumulation of superoxide
anion may also inhibit p53 protein activity in the long term,
independent of NO levels. Additionally, a significant finding of
the present study was that uric acid reduced NO levels in VSMCs
regardless of the exposure time and dose. The effects of uric acid
on protein carbonylation and lipid peroxidation in VSMCs are
little-known oxidative damage parameters that need further
investigation.
The results of the present study indicated that uric
acid may significantly increase oxidative stress in VSMCs,
suggesting that controlling uric acid levels could represent a
potential therapeutic target for preventing hypertension and
related diseases. Furthermore, it is clear that additional research
is needed to understand the role of the p53 signaling pathway in
uric acid-induced oxidative stress-mediated vascular damage and to
overcome cardiovascular diseases.
The primary aim of the present study was to provide
a detailed analysis of the cellular mechanisms involved. Therefore,
in vivo experiments and clinical studies are essential to
translate these findings into real-world applications. In future
studies, further analyses, including assessing the activities of
antioxidant enzymes such as SOD, catalase and glutathione
peroxidase, may provide more comprehensive data on the effects of
uric acid, and these additional markers will help to elucidate
cellular antioxidant defense mechanisms and their response to uric
acid-induced oxidative stress. The integration of such data with
the current study could provide a more comprehensive understanding
of the effects of uric acid on vascular health.
In conclusion, while the current study lays the
groundwork by elucidating the cellular effects of uric acid in
VSMCs, further in vivo studies and clinical research are
required for a complete understanding of these mechanisms. The data
presented in the current study may serve as a foundation for future
investigations, and the findings could have broader implications
when integrated with additional studies. Therefore, future studies
will aim to include investigations on ECs and immune cells to
provide a comprehensive understanding of the effects of uric acid
on vascular health. These future studies are crucial for developing
effective therapeutic strategies to mitigate the adverse effects of
uric acid on vascular health.
Acknowledgements
Not applicable.
Funding
Funding: This research was supported by Akdeniz University
Scientific Research Project Unit (grant no. TSA-2018-3543).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SD was involved in data curation, formal analysis,
investigation, methodology and the use of software. SD also
contributed to the writing of the original draft, and participated
in the review and editing process. EY contributed to data curation,
formal analysis and methodology, as well as writing the original
draft. AY was responsible for conceptualization, funding
acquisition, investigation, project administration and providing
resources. Additionally, AY provided supervision and validation,
and was involved in the review and editing of the manuscript. SD
and AY confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were performed according to
the Guide for the Care and Use of Laboratory Animals following
experimental protocols approved by the Local Committee on Animal
Research Ethics at Akdeniz University (approval no.
727/2018.01.024).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Becker BF: Towards the physiological
function of uric acid. Free Radic Biol Med. 14:615–631.
1993.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bardin T and Richette P: Definition of
hyperuricemia and gouty conditions. Curr Opin Rheumatol.
26:186–191. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dalbeth N, Gosling A, Gaffo A and Abhishek
A: Gout. Lancet. 397:1843–1855. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Johnson RJ, Kang DH, Feig D, Kivlighn S,
Kanellis J, Watanabe S, Tuttle KR, Rodriguez-Iturbe B,
Herrera-Acosta J and Mazzali M: Is there a pathogenetic role for
uric acid in hypertension and cardiovascular and renal disease?
Hypertension. 41:1183–1190. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Feig DI, Kang DH and Johnson RJ: Uric acid
and cardiovascular risk. N Engl J Med. 359:1811–1821.
2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lanaspa MA, Sanchez-Lozada LG, Choi YJ,
Cicerchi C, Kanbay M, Roncal-Jimenez CA, Ishimoto T, Li N, Marek G,
Duranay M, et al: Uric acid induces hepatic steatosis by generation
of mitochondrial oxidative stress: Potential role in
fructose-dependent and-independent fatty liver. J Biol Chem.
287:40732–40744. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sautin YY, Nakagawa T, Zharikov S and
Johnson RJ: Adverse effects of the classic antioxidant uric acid in
adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am
J Physiol Cell Physiol. 293:C584–C596. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ames BN, Cathcart R, Schwiers E and
Hochstein P: Uric acid provides an antioxidant defense in humans
against oxidant- and radical-caused aging and cancer: A hypothesis.
Proc Natl Acad Sci USA. 78:6858–6862. 1981.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Peden DB, Hohman R, Brown ME, Mason RT,
Berkebile C, Fales HM and Kaliner MA: Uric acid is a major
antioxidant in human nasal airway secretions. Proc Natl Acad Sci
USA. 87:7638–7642. 1990.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sautin YY and Johnson RJ: Uric acid: The
oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids.
27:608–619. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhao G, Huang L, Song M and Song Y:
Baseline serum uric acid level as a predictor of cardiovascular
disease related mortality and all-cause mortality: A meta-analysis
of prospective studies. Atherosclerosis. 231:61–68. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang W, Iso H, Murakami Y, Miura K, Nagai
M, Sugiyama D, Ueshima H and Okamura T: EPOCH-JAPAN GROUP. Serum
uric acid and mortality form cardiovascular disease: EPOCH-JAPAN
study. J Atheroscler Thromb. 23:692–703. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lee SW, Kim HC, Nam C, Lee HY, Ahn SV, Oh
YA and Suh I: Age-differential association between serum uric acid
and incident hypertension. Hypertens Res. 42:428–437.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sakata S, Hata J, Honda T, Hirakawa Y,
Oishi E, Shibata M, Yoshida D, Goto K, Kitazono T and Ninomiya T:
Serum uric acid levels and cardiovascular mortality in a general
Japanese population: The Hisayama study. Hypertens Res. 43:560–568.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kurra V, Vehmas T, Eräranta A, Jokihaara
J, Pirttiniemi P, Ruskoaho H, Tokola H, Niemelä O, Mustonen J and
Pörsti I: Effects of oxonic acid-induced hyperuricemia on
mesenteric artery tone and cardiac load in experimental renal
insufficiency. BMC Nephrol. 16(35)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Garcia-Arroyo FE, Gonzaga G, Munoz-Jimenez
I, Blas-Marron MG, Silverio O, Tapia E, Soto V, Ranganathan N,
Ranganathan P, Vyas U, et al: Probiotic supplements prevented
oxonic acid-induced hyperuricemia and renal damage. PLoS One.
13(e0202901)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mazzali M, Hughes J, Kim YG, Jefferson JA,
Kang DH, Gordon KL, Lan HY, Kivlighn S and Johnson RJ: Elevated
uric acid increases blood pressure in the rat by a novel
crystal-independent mechanism. Hypertension. 38:1101–1106.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yu MA, Sánchez-Lozada LG, Johnson RJ and
Kang DH: Oxidative stress with an activation of the
renin-angiotensin system in human vascular endothelial cells as a
novel mechanism of uric acid-induced endothelial dysfunction. J
Hypertens. 28:1234–1242. 2010.PubMed/NCBI
|
|
19
|
Hsu WL, Li SY, Liu JS, Huang PH, Lin SJ,
Hsu CC, Lin YP and Tarng DC: High uric acid ameliorates indoxyl
sulfate-induced endothelial dysfunction and is associated with
lower mortality among hemodialysis patients. Toxins (Basel).
9(20)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cai W, Duan XM, Liu Y, Yu J, Tang YL, Liu
ZL, Jiang S, Zhang CP, Liu JY and Xu JX: Uric acid induces
endothelial dysfunction by activating the HMGB1/RAGE signaling
pathway. Biomed Res Int. 2017(4391920)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ko J, Kang HJ, Kim DA, Kim MJ, Ryu ES, Lee
S, Ryu JH, Roncal C, Johnson RJ and Kang DH: Uric acid induced the
phenotype transition of vascular endothelial cells via induction of
oxidative stress and glycocalyx shedding. FASEB J. 33:13334–13345.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sandoo A, van Zanten JJ, Metsios GS,
Carroll D and Kitas GD: The endothelium and its role in regulating
vascular tone. Open Cardiovasc Med J. 4:302–312. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Brozovich F, Nicholson C, Degen C, Gao YZ,
Aggarwal M and Morgan K: Mechanisms of vascular smooth muscle
contraction and the basis for pharmacologic treatment of smooth
muscle disorders. Pharmacol Rev. 68:476–532. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Touyz RM, Alves-Lopes R, Rios FJ, Camargo
LL, Anagnostopoulou A, Arner A and Montezano AC: Vascular smooth
muscle contraction in hypertension. Cardiovasc Res. 114:529–539.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Radi R: Oxygen radicals, nitric oxide, and
peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad
Sci USA. 115:5839–5848. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Maruhashi T, Hisatome I, Kihara Y and
Higashi Y: Hyperuricemia and endothelial function: From molecular
background to clinical perspectives. Atherosclerosis. 278:226–231.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yu W and Cheng JD: Uric acid and
cardiovascular disease: An update from molecular mechanism to
clinical perspective. Front Pharmacol. 11(582680)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pacher P, Obrosova IG, Mabley JG and Szabó
C: Role of nitrosative stress and peroxynitrite in the pathogenesis
of diabetic complications. Emerging new therapeutical strategies.
Curr Med Chem. 12:267–275. 2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang F, Yuan Q, Chen F, Pang J, Pan C, Xu
F and Chen Y: Fundamental mechanisms of the cell death caused by
nitrosative stress. Front Cell Dev Biol. 9(742483)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Choi YJ, Yoon Y, Lee KY, Hien TT, Kang KW,
Kim KC, Lee J, Lee MY, Lee SM, Kang DH and Lee BH: Uric acid
induces endothelial dysfunction by vascular insulin resistance
associated with the impairment of nitric oxide synthesis. FASEB J.
28:3197–3204. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mishima M, Hamada T, Maharani N, Ikeda N,
Onohara T, Notsu T, Ninomiya H, Miyazaki S, Mizuta E, Sugihara S,
et al: Effects of uric acid on the NO production of HUVECs and its
restoration by urate lowering agents. Drug Res (Stuttg).
66:270–274. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lin Y, Xie Y, Hao Z, Bi H, Liu Y, Yang X
and Xia Y: Protective effect of uric acid on ox-LDL-induced HUVECs
injury via Keap1-Nrf2-ARE pathway. J Immunol Res.
2021(5151168)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ouyang R, Zhao X, Zhang R, Yang J, Li S
and Deng D: FGF21 attenuates high uric acid-induced endoplasmic
reticulum stress, inflammation and vascular endothelial cell
dysfunction by activating Sirt1. Mol Med Rep. 25(35)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Li P, Zhang L, Zhang M, Zhou C and Lin N:
Uric acid enhances PKC-dependent eNOS phosphorylation and mediates
cellular ER stress: A mechanism for uric acid-induced endothelial
dysfunction. Int J Mol Med. 37:989–997. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huang Z, Hong Q, Zhang X, Xiao W, Wang L,
Cui S, Feng Z, Lv Y, Cai G, Chen X and Wu D: Aldose reductase
mediates endothelial cell dysfunction induced by high uric acid
concentrations. Cell Commun Signal. 15(3)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lee TS, Lu TM, Chen CH, Guo BC and Hsu CP:
Hyperuricemia induces endothelial dysfunction and accelerates
atherosclerosis by disturbing the asymmetric
dimethylarginine/dimethylarginine dimethylaminotransferase 2
pathway. Redox Biol. 46(102108)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Corry DB, Eslami P, Yamamoto K, Nyby MD,
Makino H and Tuck ML: Uric acid stimulates vascular smooth muscle
cell proliferation and oxidative stress via the vascular
renin-angiotensin system. J Hypertens. 26:269–275. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Doğru S, Yaşar E and Yeşilkaya A: Uric
acid can enhance MAPK pathway-mediated proliferation in rat primary
vascular smooth muscle cells via controlling of mitochondria and
caspase-dependent cell death. J Recept Signal Transduct Res.
42:293–301. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li T, Kon N, Jiang L, Tan M, Ludwig T,
Zhao Y, Baer R and Gu W: Tumor suppression in the absence of
p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell.
149:1269–1283. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Itahana Y and Itahana K: Emerging roles of
p53 family members in glucose metabolism. Int J Mol Sci.
19(776)2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mercer J and Bennett M: The role of p53 in
atherosclerosis. Cell Cycle. 5:1907–1909. 2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Phadwal K, Tang QY, Luijten I, Zhao JF,
Corcoran B, Semple RK, Ganley IG and MacRae VE: p53 regulates
mitochondrial dynamics in vascular smooth muscle cell
calcification. Int J Mol Sci. 24(1643)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Popowich DA, Vavra AK, Walsh CP,
Bhikhapurwala HA, Rossi NB, Jiang Q, Aalami OO and Kibbe MR:
Regulation of reactive oxygen species by p53: Implications for
nitric oxide-mediated apoptosis. Am J Physiol Heart Circ Physiol.
298:H2192–H2200. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Itahana Y, Han R, Barbier S, Lei Z, Rozen
S and Itahana K: The uric acid transporter SLC2A9 is a direct
target gene of the tumor suppressor p53 contributing to antioxidant
defense. Oncogene. 34:1799–1810. 2015.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kang DH, Han L, Ouyang X, Kahn AM,
Kanellis J, Li P, Feng L, Nakagawa T, Watanabe S, Hosoyamada M, et
al: Uric acid causes vascular smooth muscle cell proliferation by
entering cells via a functional urate transporter. Am J Nephrol.
25:425–433. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Oğuz N, Kırça M, Çetin A and Yeşilkaya A:
Effect of uric acid on inflammatory COX-2 and ROS pathways in
vascular smooth muscle cells. J Recept Signal Transduct Res.
37:500–505. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Tang L, Xu Y, Wei Y and He X: Uric acid
induces the expression of TNF-α via the ROS-MAPK-NF-κB signaling
pathway in rat vascular smooth muscle cells. Mol Med Rep.
16:6928–6933. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
National Research Council: Guide for the
Care and Use of Laboratory Animals: 8th edition. The National
Academies Press, Washington, DC, 2011. https://doi.org/10.17226/12910.
|
|
50
|
American Veterinary Medical Association:
AVMA Guidelines for the Euthanasia of Animals. AVMA, Schaumburg,
IL, 2020. https://www.avma.org/resources-tools/avma-policies/avma-guidelines-euthanasia-animals.
|
|
51
|
Boston University: Institutional animal
care and use committee (IACUC) Guidelines. https://www.bu.edu/research/ethics-compliance/.
|
|
52
|
University of Maryland: Animal care and
use Training. https://research.umd.edu/resources/department-laboratory-animal-resources-dlar/animal-care-and-use-training.
|
|
53
|
Ahmadi-Noorbakhsh S, Abbasi MF, Ghasemi M,
Bayat G, Davoodian N, Sharif-Paghaleh E, Poormoosavi SM, Rafizadeh
M, Maleki M, Shirzad-Aski H, et al: Anesthesia and analgesia for
common research models of adult mice. Lab Anim Res.
38(40)2022.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Parasuraman S and Christapher PV:
Anesthesia and euthanasia of experimental animals. In: Introduction
to basics of pharmacology and toxicology: Volume 3: Experimental
Pharmacology: Research methodology and biostatistics. Springer,
pp65-75, 2022.
|
|
55
|
Gunther S, Alexander RW, Atkinson WJ and
Gimbrone MA Jr: Functional angiotensin II receptors in cultured
vascular smooth muscle cells. J Cell Biol. 92:289–298.
1982.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Görlach A, Brandes RP, Bassus S, Kronemann
N, Kirchmaier CM, Busse R and Schini-Kerth VB: Oxidative stress and
expression of p22phox are involved in the up-regulation of tissue
factor in vascular smooth muscle cells in response to activated
platelets. FASEB J. 14:1518–1528. 2000.PubMed/NCBI
|
|
57
|
Muraoka S and Miura T: Inhibition by uric
acid of free radicals that damage biological molecules. Pharmacol
Toxicol. 93:284–289. 2003.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Rodrigo R, González J and Paoletto F: The
role of oxidative stress in the pathophysiology of hypertension.
Hyperten Res. 34:431–440. 2011.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liu N, Xu H, Sun Q, Yu X, Chen W, Wei H,
Jiang J, Xu Y and Lu W: The role of oxidative stress in
hyperuricemia and xanthine oxidoreductase (XOR) inhibitors. Oxid
Med Cell Longev. 2021(1470380)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Levine AB, Punihaole D and Levine TB:
Characterization of the role of nitric oxide and its clinical
applications. Cardiology. 122:55–68. 2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Napoli C, Paolisso G, Casamassimi A,
Al-Omran M, Barbieri M, Sommese L, Infante T and Ignarro LJ:
Effects of nitric oxide on cell proliferation: Novel insights. J Am
Coll Cardiol. 62:89–95. 2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Gherghina ME, Peride I, Tiglis M, Neagu
TP, Niculae A and Checherita IA: Uric acid and oxidative
stress-relationship with cardiovascular, metabolic, and renal
impairment. Int J Mol Sci. 23(3188)2022.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Li M, Qian M, Kyler K and Xu J:
Endothelial-vascular smooth muscle cells interactions in
atherosclerosis. Front Cardiovasc Med. 5(151)2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hirase T and Node K: Endothelial
dysfunction as a cellular mechanism for vascular failure. Am J
Physiol Heart Circ Physiol. 302:H499–H505. 2012.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Vanhoutte PM, Zhao Y, Xu A and Leung SW:
Thirty years of saying NO: Sources, fate, actions, and misfortunes
of the endothelium-derived vasodilator mediator. Circ Res.
119:375–396. 2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 30:H2181–H2190. 2011.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Sugamura K and Keaney JF Jr: Reactive
oxygen species in cardiovascular disease. Free Radic Biol Med.
51:978–992. 2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Drummond GR, Selemidis S, Griendling KK
and Sobey CG: Combating oxidative stress in vascular disease: NADPH
oxidases as therapeutic targets. Nat Rev Drug Discov. 10:453–471.
2011.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Murray M, Selby-Pham S, Colton BL, Bennett
L, Williamson G and Dordevic AL: Does timing of phytonutrient
intake influence the suppression of postprandial oxidative stress?
A systematic literature review. Redox Biol.
46(102123)2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Baradaran A, Nasri H and Rafieian-Kopaei
M: Oxidative stress and hypertension: Possibility of hypertension
therapy with antioxidants. J Res Med Sci. 19:358–367.
2014.PubMed/NCBI
|
|
71
|
Tahhan AS, Sandesara PB, Hayek SS,
Alkhoder A, Chivukula K, Hammadah M, Mohamed-Kelli H, O'Neal WT,
Topel M, Ghasemzadeh N, et al: Association between oxidative stress
and atrial fibrillation. Heart Rhythm. 14:1849–1855.
2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Ahmad KA, Yuan DY, Nawaz W, Ze H, Zhuo CX,
Talal B, Taleb A, Mais E and Qilong D: Antioxidant therapy for
management of oxidative stress induced hypertension. Free Radic
Res. 51:428–438. 2017.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Kattoor AJ, Pothineni NVK, Palagiri D and
Mehta JL: Oxidative stress in atherosclerosis. Curr Atheroscler
Rep. 19(42)2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Dai Y, Cao Y, Zhang Z, Vallurupalli S and
Mehta JL: Xanthine oxidase induces foam cell formation through
LOX-1 and NLRP3 activation. Cardiovasc Drugs Ther. 31:19–27.
2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Coliva G, Lange M, Colombo S, Chervet JP,
Domingues MR and Fedorova M: Sphingomyelins prevent propagation of
lipid peroxidation-LC-MS/MS evaluation of inhibition mechanisms.
Molecules. 25(1925)2020.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Chan HH, Chan E, Kwok CTK, Leung GPH, Lee
SMY and Seto SW: The role of p53 in the alternation of vascular
functions. Front Pharmacol. 13(981152)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Mercer J, Figg N, Stoneman V, Braganza D
and Bennett MR: Endogenous p53 protects vascular smooth muscle
cells from apoptosis and reduces atherosclerosis in ApoE knockout
mice. Circ Res. 96:667–674. 2005.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Cao RY, Eves R, Jia L, Funk CD, Jia Z and
Mak AS: Effects of p53-knockout in vascular smooth muscle cells on
atherosclerosis in mice. PLoS One. 12(e0175061)2017.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Wang M and Attardi LD: A balancing act:
p53 activity from tumor suppression to pathology and therapeutic
implications. Annu Rev Pathol. 17:205–226. 2022.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Kastenhuber ER and Lowe SW: Putting p53 in
context. Cell. 170:1062–1078. 2017.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Buizza L, Cenini G, Lanni C,
Ferrari-Toninelli G, Prandelli C, Govoni S, Buoso E, Racchi M,
Barcikowska M, Styczynska M, et al: Conformational altered p53 as
an early marker of oxidative stress in Alzheimer's disease. PLoS
One. 7(e29789)2012.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Yang L, Chang B, Guo Y, Wu X and Liu L:
The role of oxidative stress-mediated apoptosis in the pathogenesis
of uric acid nephropathy. Ren Fail. 41:616–622. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Liu Y and Gu W: The complexity of
p53-mediated metabolic regulation in tumor suppression. Semin
Cancer Biol. 85:4–32. 2022.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Kibbe MR, Li J, Nie S, Choi BM, Kovesdi I,
Lizonova A, Billiar TR and Tzeng E: Potentiation of nitric
oxide-induced apoptosis in p53-/- vascular smooth muscle cells. Am
J Physiol Cell Physiol. 282:C625–C634. 2002.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Kim YM, Choi BM, Kim YS, Kwon YG, Kibbe
MR, Billiar TR and Tzeng E: Protective effect of p53 in vascular
smooth muscle cells against nitric oxide-induced apoptosis is
mediated by up-regulation of heme oxygenase-2. BMB Rep. 41:164–169.
2008.PubMed/NCBI View Article : Google Scholar
|