Introduction
Tripterygium wilfordii is a traditional
Chinese medicinal herb that is commonly used for treating a number
of conditions, such as arthritis, dermatitis, lupus erythematosus
and eczema (1-4).
Previous studies have also highlighted its potential for antitumor
therapy and immune regulation (5-8).
Tripterygium wilfordii contains >100 chemical compounds,
which can be primarily categorized into alkaloids, terpenes and
triterpenes (9). The Tripterygium
polycoride tablet, a non-steroidal immunosuppressive agent
(10), was developed and introduced
for clinical use in China. This tablet has been shown to inhibit
the secretion of IL-2 and the expression of IL-2 receptors on T
lymphocytes (11), in addition to
inducing lymphocyte apoptosis whilst downregulating T cell receptor
signaling (12).
Tripterine, also known as celastrol, is an active
compound that can be extracted from Tripterygium wilfordii.
It is one of the key ingredients in the Chinese patent medicine
Tripterygium tablet (with each tablet containing ~35 µg tripterine)
(13). Numerous studies have
demonstrated its immunomodulatory and antitumor properties
(14-20).
Additionally, tripterine has been shown to regulate the expression
of various drug-metabolizing enzymes and drug transporters. Jin
et al (21) previously
examined the effects of tripterine on five subtypes of cytochrome
P450 (CYP) enzymes (CYP1A2, CYP2C19, CYP2D6, CYP2E1 and CYP3A4) in
human liver microsomes, before finding that tripterine inhibited
these enzymes to varying extents. Similarly, Sun et al
(22) reported that tripterine
inhibited CYP1A2, CYP2C11, CYP2D6, CYP2E1 and CYP3A2, acting as a
mixed-type inhibitor of CYP3A4 and a competitive inhibitor of
CYP1A2 and CYP2C11. In addition, Zhang et al (23) demonstrated that celastrol
(tripterine) is a potent inhibitor of uridine diphosphate
glucuronosyltransferase (UGT) 1A6 and UGT2B7(23). In another study, Zhao et al
(24) found that tripterine could
upregulate farnesoid X receptor (FXR) expression in the liver.
Given its potential impact on the activity of various
drug-metabolizing enzymes and transporters, tripterine has the
potential to exert herb-drug interactions. Furthermore, tripterine
has been shown to inhibit intestinal lipid absorption (25). Since bile acids and dietary fats
serve a significant role in the oral bioavailability of
cyclosporine A (CsA) (26,27), any changes in bile acid circulation
and fat absorption induced by tripterine may notably influence the
absorption and pharmacokinetics of orally administered drugs.
CsA is one of the most commonly used
immunosuppressive agents in clinical practice, though it has a
narrow therapeutic window. Numerous studies have suggested a
potential for the combined use of Tripterygium wilfordii and
CsA. Yang et al (28)
reported that CsA combined with Tripterygium glycosides could
prolong the mean survival time and decrease the rate of allograft
rejection of the cardiac allografts. Additionally, Tripterygium
glycosides have shown a synergistic effect with CsA for the
treatment of immune rejection following organ transplantation
(29-32).
This synergy is considered to be associated with the
immunomodulatory effects of Tripterygium glycosides.
CsA is a substrate of CYP3A, p-glycoprotein (P-gp),
UGT1A, and multidrug resistance protein (MRP) 2 (33-36).
Additionally, because CsA is a fat-soluble macromolecule, its oral
bioavailability is significantly influenced by bile secretion and
intestinal lipid absorption (37).
Provided that Tripterine has been shown to affect drug-metabolizing
enzymes and transporters, it is possible that it may alter the
pharmacokinetics of CsA. However, to the best of our knowledge, the
specific effects of Tripterine on CsA have not been reported to
date.
Therefore, in the present study, the aim was to
investigate the impact of Tripterine on the pharmacokinetics of CsA
in rats. After 7 consecutive days of Tripterine pretreatment, CsA
was orally administered, before the whole blood concentration of
CsA was periodically measured. The transcriptional and protein
expression levels of several bile-related transporters, drug
transporters, metabolic enzymes and nuclear receptors were also
assessed to explore the underlying mechanisms, using previous
developed experimental methods (38).
Materials and methods
Materials
Tripterine, CsA and cyclosporine D (CsD) were
purchased from Dalian Meilun Biology Technology Co., Ltd.
Animals
A total of 36 male Sprague-Dawley (SD) rats (weight,
180-220 g; age, 6-8 weeks) were purchased from the Laboratory
Animal Research Center of Tongji Medical College of Huazhong
University of Science and Technology (Wuhan, China) and were given
access to a commercial rat chow diet and tap water. The animals
were housed, two per cage and were maintained at 22±2˚C and 50-60%
relative humidity, under a 12-h light/dark cycle. The experiments
were initiated after acclimation under these conditions for ≥1
week. The experiments were performed in accordance with the
‘Guiding Principles in the Use of Animals in Toxicology’ adopted by
the Society of Toxicology (USA) in July 1989 and revised in March
1999. All animal studies were approved (approval no. 4352; date,
2023) by The Institutional Animal Care and Use Committee of Tongji
Medical College Huazhong University of Science and Technology
(Wuhan, China).
Pharmacokinetic studies in rats
Experiment one. In total, 24 rats were
randomly divided into the following 4 groups (n=6): i) Control
group, which was pretreated with 0.5% Carboxymethylcellulose sodium
(CMC-Na); and ii) tripterine pretreated groups (6, 18 and 54 mg/kg,
respectively). Tripterine were all prepared with the corresponding
standard and 0.5% CMC-Na as suspension, which were shaken well
before gavage. Rats in the control group were administered 0.5%
CMC-Na by gavage. All groups of rats were administered by oral
gavage for 7 consecutive days. The rats were fasted for ≥12 h
(overnight) before the day of experiment and drank freely. On the
experimental day (day 7), 30 min after the last dose of 0.5% CMC-Na
or tripterine, CsA (10 mg/kg) was administered to the rats by oral
gavage. Blood samples were collected at 1, 2, 4, 5, 6, 7, 8, 12,
24, 36, 48 and 72 h after the administration of CsA. All the blood
samples were stored at -80˚C until analysis.
Experiment two. Rats were randomly divided
into 4 groups consistent with experiment one, but there were only 3
rats in each group. All of these rats were administered by oral
gavage for 7 days. The rats were fasted for ≥12 h (overnight)
before the day of experiment and drank freely. After 2 h of
intragastric administration on day 7, the rats were euthanized by
injection of 100 mg/kg sodium pentobarbital. The liver, kidney and
mucosa of small intestine were isolated, washed with saline and
blotted dry. The samples were stored at -80˚C until further
use.
Quantification of CsA by liquid
chromatography-mass spectrometry (MS)/MS (LC-MS/MS)
The method for the determination of CsA was based on
a previously developed l LC-MS)/MS method (39). The linear concentration range of CsA
was 5-4,000 ng/ml and the lower limit of quantification was 5
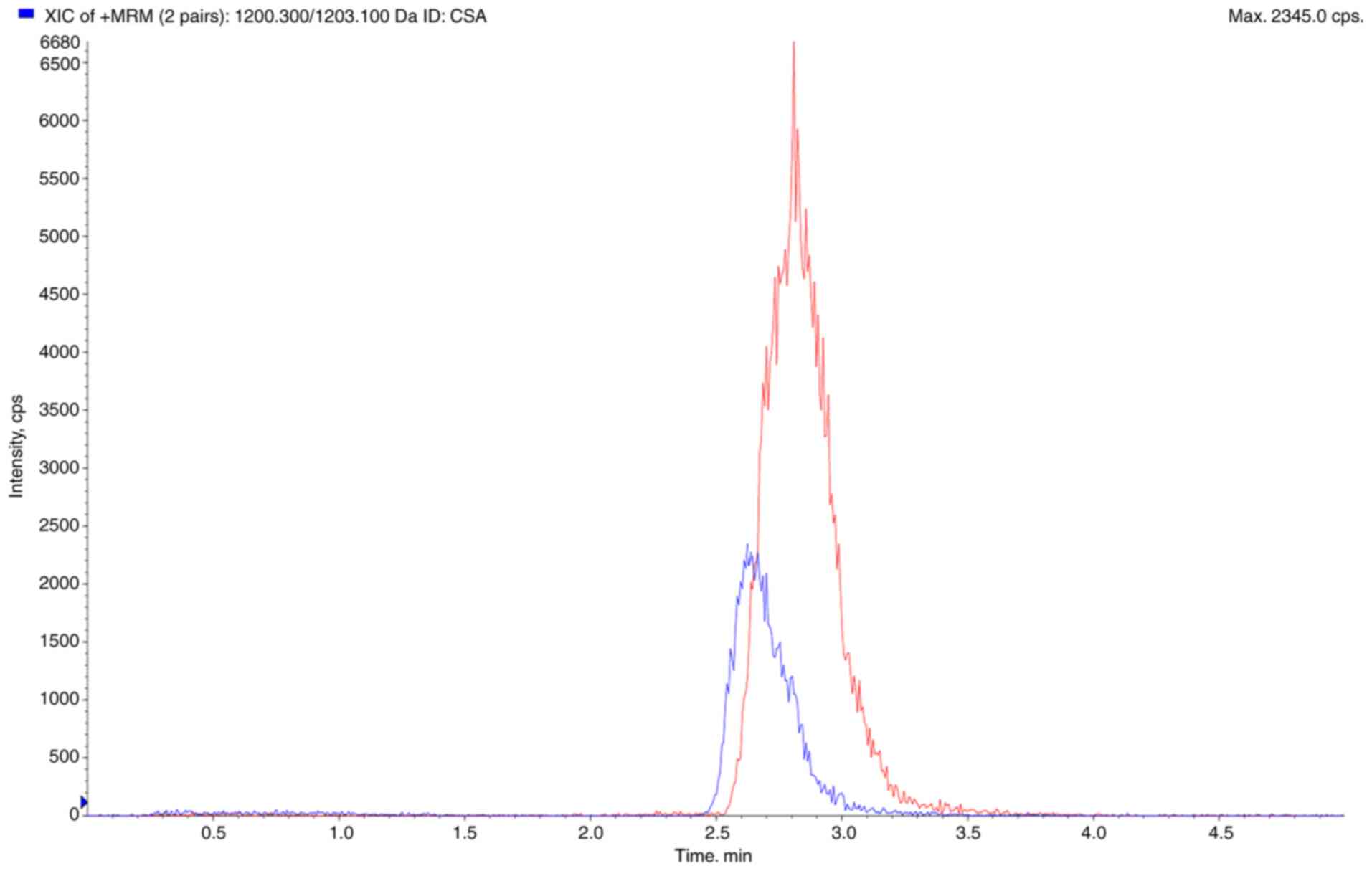
ng/ml. The extracted ion chromatogram diagram of CSA and CSD is
shown in Fig. 1.
Sample preparation. A total of 100 µl of
whole blood was absorbed into a 1.5-ml EP tube, vortexed for 5 min
with 10 µl methanol, 300 µl water, 240 µl of 200 ng/ml CsD, and 60
µl of 0.4 mol/l ZnSO4. The mixture was centrifuged at 21,012.8 x g
for 5 min, and the supernatant was collected for analysis.
HPLC-MS/MS. LC separation and MS detection
were performed using the AB Sciex API 4000™ LC/MS/MS system.
Chromatographic separation was performed on an ACE excel 5 C18
column (50x2.1 mm). The mobile phase consisted of 10 mmol/l
ammonium acetate in aqueous solution (A) and Acetonitrile (B),
(containing 0.1% formic acid), at a total flow rate of 0.4 ml/min.
The gradient profile for the LC pumps under the final
chromatography conditions were as follows: 0 min, 50:50; 0.1-3 min,
0:100; 3.1-5 min, 50:50 (A:B, v/v) (Fig. 1). The injection volume of all
samples was 10 µl. The column temperature was set at 40˚C, and the
sample tray temperature was maintained at 4˚C. For MS detection,
the ESI source was operated in the negative ion mode. High purity
nitrogen was used as the sheath (35 arb) and auxiliary (15 arb) gas
and high-purity argon was used as the collision gas (1.5 mTorr).
The parameters were as follows: spray voltage, 5.5 kV; capillary
temperature, 300˚C; scan width for SRM, 0.5 m/z; scan time, 0.2
sec. The peak width settings for both Q1 and Q3 were 0.7 m/z. The
SRM ion pair transitions and collision energy levels of each
component are listed in the Table
SI.
Calibration curves. CSA was dissolved in
methanol and prepared into a 1 mg/ml stock solution. The stock
solution was diluted in methanol gradient to working standard
solutions of 40,000, 20,000, 10,000, 5,000, 1,600, 400, 100 and 50
ng/ml. For analyte identification and quantification, calibration
standards were prepared by spiking 10 µl of working standard
solutions into 100 µl of blank rat blood at final concentrations of
5-4,000 ng/ml. The solutions for calibration curves were pretreated
according to the method in 1.1. Regression line of calibration
curve was y=0.000411x+0.0294, R2=0.9946.
Pharmacokinetic analysis
The blood concentration data were analyzed using the
non-compartmental method using Drug and Statistics software
(version 3.2.8; Shanghai BioGuider Medicinal Technology Co. Ltd.;
http://www.gooddrug.net/). The peak blood
concentration (Cmax) and time to reach Cmax
(Tmax) of CsA were acquired directly from the
concentration-time curve. The elimination rate constant
(Kel) was calculated using the log-linear regression of
the phase-eliminated data. The area under the plasma
concentration-time curve (AUC0-t) from time 0 to the
time of last measured concentration (Clast) was
calculated using the linear trapezoidal rule. The AUC 0 to infinity
(AUC0-∞) was obtained by the addition of
AUC0-t and the extrapolated area determined by
Clast/Kel. The terminal half-life
(T1/2) was calculated using the
formula 0.693/Kel. The mean residence time was
calculated using the curve area under the first moment vs. time
curve/AUC. Apparent clearance was calculated using the formula
Dose/AUC0-∞ and the apparent volume of distribution was
calculated using the formula CL/Kel.
Measurement of mRNA expression
Reverse transcription-quantitative PCR was used to
quantify the mRNA expression of breast cancer resistance protein
(BCRP), bile salt export pump (BSEP), constitutive
androstane receptor (CAR), CYP3A1, CYP3A2, FXR, MRP2,
Na+-taurocholate co-transporting polypeptide
(NTCP), organic anion-transporting polypeptide 1B2
(OATP1B2), P-gp, pregnane X receptor (PXR) and
UCT1A1 in the liver, the mRNA expression of BCRP, CAR,
CYP3A1, CYP3A2, FXR, MRP2, P-gp, PXR and UGT1A1 in the
small intestine and the mRNA expression of BCRP, MRP2 and
P-gp in the kidney. In total, ~100 mg tissue was received
and thoroughly ground in 1 ml pre-chilled TRIzol (Thermo Fisher
Scientific, Inc.). RNA was extracted with 250 µl chloroform,
precipitated with isopropanol and washed with 75% ethanol.
According to the manufacturer's instructions, RNA was reverse
transcribed to cDNA using the PrimeScript RT reagent kit with gDNA
Eraser (cat. no. RR047A; Takara Biotechnology Co., Ltd.). qPCR was
performed using SYBR® Premix Ex Taq™ (Takara
Biotechnology Co., Ltd.) and a StepOne Real-Time PCR System (Thermo
Fisher Scientific, Inc.). The reaction procedure involved first
pre-denaturing at 95˚C for 1 min, followed by 40 cycles of 15 sec
at 95˚C, 20 sec at 58˚C, 45 sec at 72˚C and finally the temperature
rose from 60 to 95˚C at a rate of 1˚C per 20 sec. The
2-ΔΔCq method was used to calculate the relative mRNA
expression levels (40). The
sequences of all the primers used for PCR were provided in Table SII.
Measurement of protein expression
The protein expression of BCRP (1:1,000; cat. no.
ab207732), BSEP (1:3,000; cat. no. ab217532), CAR (1:1,000; cat.
no. ab186869), CYP3A1 (1:5,000; cat. no. ab22733), CYP3A2 (1:5,000;
cat. no. ab195627), FXR (1:1,000; cat. no. ab228949), MRP2
(1:5,000; cat. no. ab203397), NTCP (1:5,000; cat. no. ab131084),
OATP1B2 (1:500; ab15442), P-gp (1:6,000; cat. no. ab170904), PXR
(1:1,000; cat. no. ab118336) and UCT1A1 (1:6,000; cat. no.
ab194697; all from Abcam) in the liver, the protein expression of
BCRP, CAR, CYP3A1, CYP3A2, FXR, MRP2, P-gp, PXR and UGT1A1 in the
small intestine and the protein expression of BCRP, MRP2 and P-gp
in the kidney of rats were analyzed by western blotting. After the
total protein in the tissues was extracted using a RIPA lysis
buffer (cat. no. G2002; Wuhan Servicebio Co., Ltd.) containing PMSF
(cat. no. G2008; Wuhan Servicebio Co., Ltd.), and the protein
concentration was measured by BCA, uniformly diluted to 20 µg/ml,
and 10 µl was loaded per well. The protein samples were separated
by 8-20% SDS-PAGE and then transferred onto PVDF membranes
(MilliporeSigma). The PVDF membranes were added to the blocking
solution (5% w/v skim milk) for 1 h at room temperature and then
incubated with the diluted primary antibodies overnight at 4˚C.
After the membranes were washed three times with TBST (TBS with 10%
Tween), the membranes were incubated with the diluted
HRP-conjugated secondary antibodies goat anti-rabbit IgG H&L
(1:10,000; cat. no. ab205718,) and goat anti-mouse IgG H&L
(1:10,000; cat. no. ab205719; both from Abcam) for 30 min at room
temperature. The freshly prepared ECL mixed solution (BeyoECL Moon;
purchased from Beyotime Institute of Biotechnology) was added
dropwise to the protein side of the membranes and exposed in a dark
room. The exposure conditions were adjusted according to different
light intensities and then development and fixing were performed.
Finally, the film was scanned and archived, before the AlphaEaseFC
software (Version 3.0; ProteinSimple) processing system was used to
analyze the optical density of the target band.
Statistical analysis
Experimental data were expressed as mean ± SD.
Statistical analysis was performed using GraphPad Prism 5.0
software (GraphPad Software; Dotmatics). P<0.05 was considered
to indicate a statistically significant difference by one-way ANOVA
followed by Tukey's HSD test as a post hoc analysis.
Results
Effect of tripterine on the
pharmacokinetics of CsA
Changes in the pharmacokinetic parameters of CsA
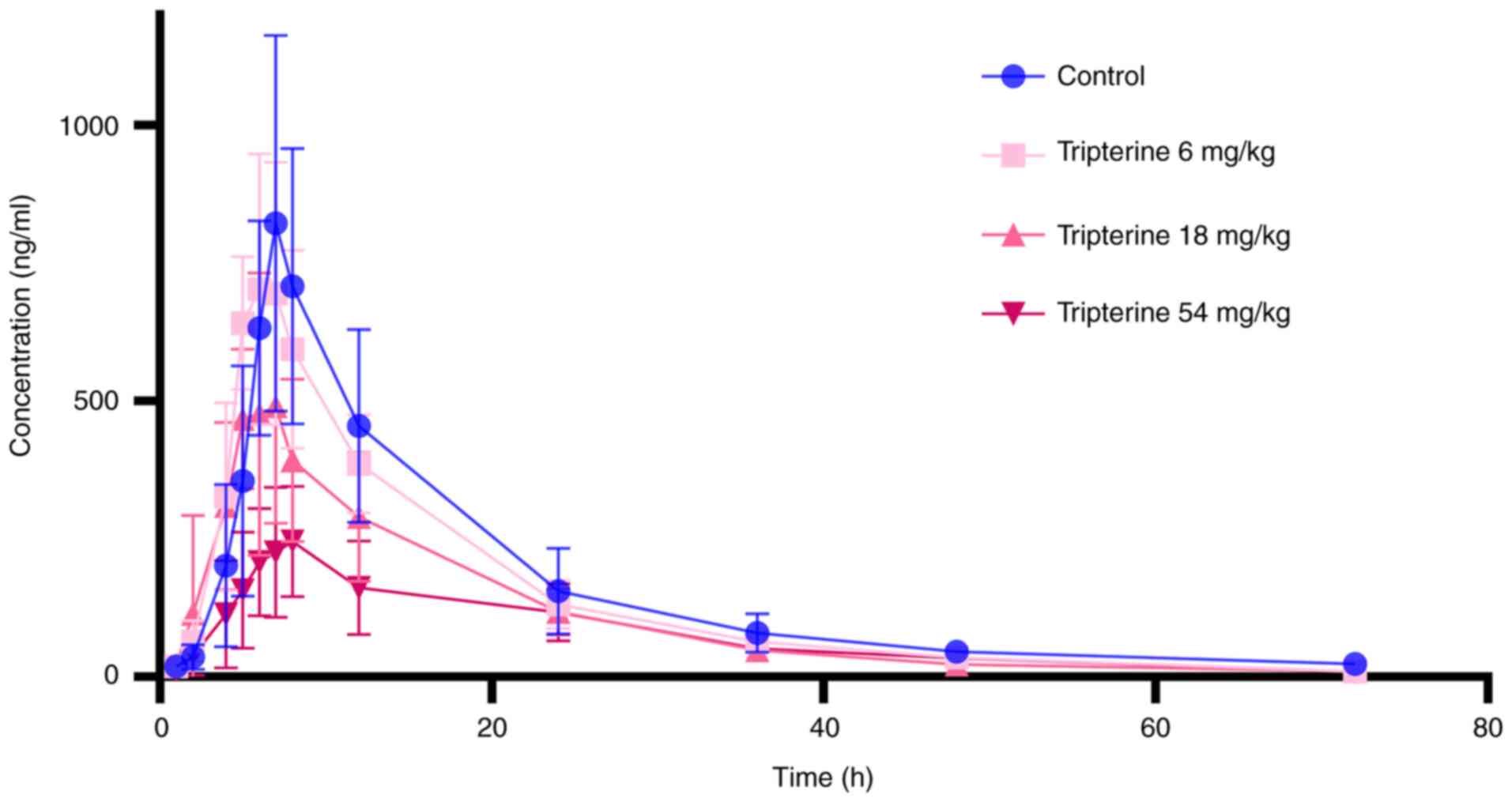
under the influence of tripterine are revealed in Fig. 2 and Table I. Tripterine exerted a
dose-dependent inhibitory effect on the blood concentration of CsA
(Fig. 2). Compared with those in
the control group, AUC0-t, AUC0-∞ and
Cmax of CsA were all found to be reduced at all doses of
tripterine administration groups. Specifically, the
AUC0-t, AUC0-∞ and Cmax of CsA
were decreased by 52.52 (P<0.05), 37.53 (P<0.05) and 66.74%
(P<0.05), respectively, whereas the CLz/F of CsA was increased
by 105.44% (P<0.05) in the 54 mg/kg dose group. Additionally,
when the dosage of tripterine was 18 mg/kg, the AUC0-∞
of CsA decreased by 33.34% (P<0.05). There was no statistically
significant difference in the other data among the administration
groups and the control group.
 | Table IPharmacokinetic parameters of CsA
after intragastric administration of CsA (10 mg/kg) in rats
pre-treated with saline (control) or tripterine (n=6). |
Table I
Pharmacokinetic parameters of CsA
after intragastric administration of CsA (10 mg/kg) in rats
pre-treated with saline (control) or tripterine (n=6).
| Parameters | Control | Triperine 6
mg/kg | Tripterine 18
mg/kg | Tripterine 54
mg/kg |
|---|
| AUC (0-t) (µg/l x
h) |
11,534.533±3,905.446 |
10,312.868±1,738.357 |
7,907.875±2,450.828 |
5,476.198±1,663.259a |
| AUC (0-∞) (µg/l x
h) |
1,2014.629±3,724.995 |
10,473.354±1,714.618 |
8,008.644±2470.771a |
5,705.554±1,624.031a |
| Mean residence time
(0-t) (h) | 18.16±2.19 | 16.38±1.14 | 16.502±1.167 | 22.112±2.956 |
| t1/2z (h) | 18.995±8.132 | 12.4±2.1 | 11.77±2.95 | 15.86±7.006 |
| Tmax (h) | 7.167±0.408 | 6.167±1.169 | 6.167±1.472 | 7±1.265 |
| Vz/F (l/kg) | 28.537±24.462 | 17.799±5.386 | 23.422±10.932 |
47.16±38.51a |
| CLz/F (l/h/kg) | 0.919±0.347 | 0.98±0.19 | 1.362±0.446 |
1.888±0.593a |
| Cmax (µg/l) | 827±331.162 |
794.833±249.397 | 594.83±222.34 |
275±114a |
Relative mRNA expression levels of
drug-metabolizing enzymes (DMEs), drug transporters (DTs) and
nuclear receptors (NRs) in the liver, kidney and small intestine
after the treatment of tripterine
The mRNA levels of DMEs, DTs and NRs were determined
by RT-qPCR (Fig. 3).
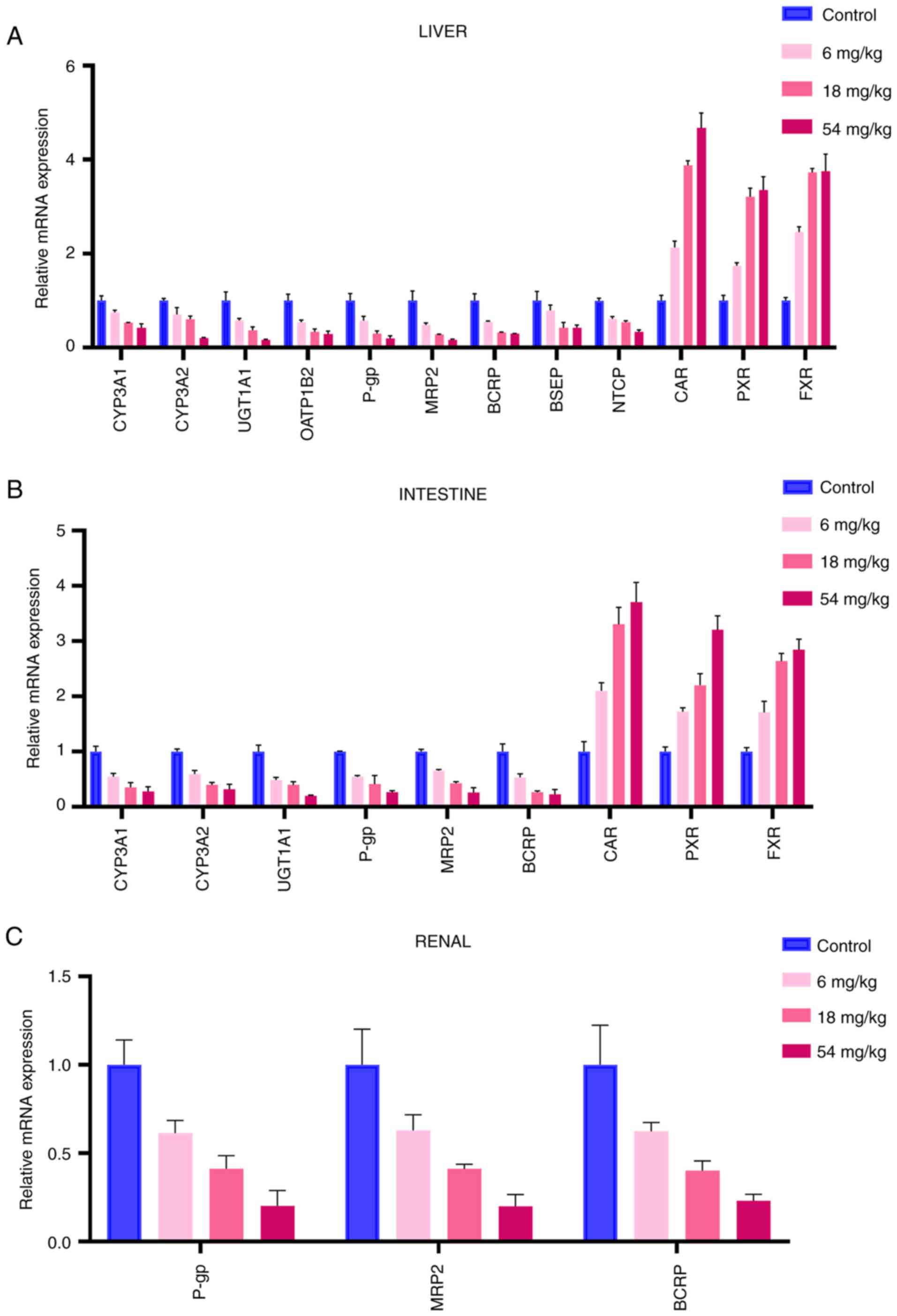 | Figure 3Effect of tripterine on the mRNA
expression levels. (A) Effect of tripterine on the mRNA expression
levels of CYP3A1, CYP3A2, UGT1A1, OATP1B2, P-gp, BCRP, MRP2, BSEP,
NTCP, CAR, PXR and FXR in the liver. (B) Effect of tripterine on
the mRNA expression levels of CYP3A1, CYP3A2, UGT1A1, P-gp, BCRP,
MRP2, CAR, PXR and FXR in the intestine. (C) Effect of tripterine
on the mRNA expression levels of P-gp, BCRP and MRP2 in the renal
tissues. Relative mRNA expression levels in rats in the control and
different doses of tripterine groups were measured by reverse
transcription-quantitative PCR and calculated as comparative levels
over control using the 2-∆∆Cq method. Vertical bars
represent the mean ± SD (n=3). CYP, Cytochrome P450 proteins; UGT,
UDP-glucuronosyltransferases; OATP, organic anion transporting
polypeptides; P-gp, P-glycoprotein; BCRP, breast cancer resistance
protein; MRP, multi-drug resistance protein; BSEP, bile salt export
pump; NTCP, sodium taurocholate co-transporting polypeptide; CAR,
constitutive androstane receptor; PXR, pregnane X receptor; FXR,
farnesoid X receptor. |
mRNA expression levels of DMEs, DTs
and NRs in the liver
As demonstrated in Fig.
3A, the mRNA expression of CYP3A1, CYP3A2, UGT1A1, OATP1B2,
P-gp, MRP2, BCRP, BSEP, NTCP, CAR, PXR and FXR in the liver was
measured. The mRNA expression of DMEs and DTs was found to be
decreased, whilst the expression of NRs was increased, compared
with those in the control group. The mRNA expression levels of
CYP3A1 in the three tripterine pre-treated groups (6, 18 and 54
mg/kg) were decreased by 24.92, 46.62 and 57.54%, respectively. The
decrease in the mRNA expression level of CYP3A2 in the 6, 18 and 54
mg/kg tripterine pre-treated groups was 29.26, 39.42 and 79.25%,
respectively. In addition, the mRNA expression level of UGT1A1 in
the 6, 18 and 54 mg/kg tripterine pre-treated groups was decreased
by 41.82, 62.6 and 84.04%, respectively. The mRNA expression level
of OATP1B2 in the 6, 18 and 54 mg/kg tripterine pre-treated groups
was decreased by 45.78, 66.67 and 71.46%, respectively. The
reductions of the mRNA expression level of P-gp in the 6, 18 and 54
mg/kg tripterine pre-treated groups was by 42.81, 70.09 and 80.59%,
respectively. The mRNA expression level of BCRP in the 6, 18 and 54
mg/kg tripterine pre-treated groups was decreased by 44.63, 67.61
and 70.54%, respectively. For MRP2, the expression of mRNA was
reduced by 52.03, 71.52 and 83.08% in the 6, 18 and 54 mg/kg
tripterine pre-treated groups, respectively. BSEP mRNA expression
was decreased by 20.49, 58.16 and 57.39%, respectively, in the 6,
18 and 54 mg/kg pre-treatment groups. The mRNA expression level of
NTCP in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
decreased by 38.46, 46.04 and 66.12%, respectively. The mRNA
expression level of CAR in the 6, 18 and 54 mg/kg tripterine
pre-treated groups was increased by 113.5, 287.7 and 367.68%,
respectively. The mRNA expression level of PXR in the 6, 18 and 54
mg/kg tripterine pre-treated groups was raised by 74.04, 221.33 and
235.20%, respectively. The increase of FXR mRNA expression in the
6, 18 and 54 mg/kg dose groups was 145.4, 272.55 and 275.28%,
respectively.
mRNA expression levels of DMEs, DTs
and NRs in the intestine
As revealed in Fig.
3B, the mRNA expression of CYP3A1, CYP3A2, UGT1A1, P-gp, MRP2,
BCRP, CAR, PXR and FXR in the intestine was measured. The mRNA
expression level of CYP3A1 in three tripterine pretreated groups
(6, 18 and 54 mg/kg) was decreased by 45.07, 64.55 and 71.89%,
respectively. The decrease in the mRNA expression level of CYP3A2
in the 6, 18 and 54 mg/kg tripterine pre-treated groups was 40.48,
60.33 and 67.96%, respectively. The mRNA expression level of UGT1A1
in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
decreased by 41.82, 62.60 and 84.04%, respectively. The reductions
of the mRNA expression level of P-gp in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was by 42.81, 70.09 and 80.59%. The
mRNA expression level of BCRP in the 6, 18 and 54 mg/kg tripterine
pre-treated groups was decreased by 46.4, 73.69 and 77.22%,
respectively. In terms of MRP2, the expression of its mRNA was
reduced by 37.03, 58.88 and 80.06% in the 6, 18 and 54 mg/kg
tripterine pre-treated groups. The mRNA expression level of CAR in
the 6, 18 and 54 mg/kg tripterine pre-treated groups was increased
by 110.16, 230.32 and 270.74% respectively. The mRNA expression
level of PXR in the 6, 18 and 54 mg/kg tripterine pre-treated
groups was raised by 72.16, 120.29 and 220.24%. The increase of FXR
mRNA expression in the 6, 18 and 54 mg/kg tripterine pre-treated
groups was 71.02%, 163.56 and 184.31%, respectively.
mRNA expression levels of DTs in the
kidney
As shown in Fig. 3C,
the mRNA expression of P-gp, MRP2 and BCRP was measured in the
kidney. The mRNA expression level of P-gp in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was decreased by 38.6, 58.7 and
79.73%, respectively. The mRNA expression level of BCRP in the 6,
18 and 54 mg/kg tripterine pre-treated groups was decreased by
37.52, 59.82 and 76.82%, respectively. The mRNA expression level of
MRP2 in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
decreased by 39.5, 67.92 and 80.93%, respectively.
Protein expression levels of DMEs, DTs
and NRs in the liver, kidney and small intestine after the
treatment of tripterine
The protein expression of DMEs, DTs and NRs were
next determined by western blotting. Densitometric analysis results
are revealed in Fig. 4. The western
blotting images of proteins in liver, kidney and small intestine
are presented in Fig. 5.
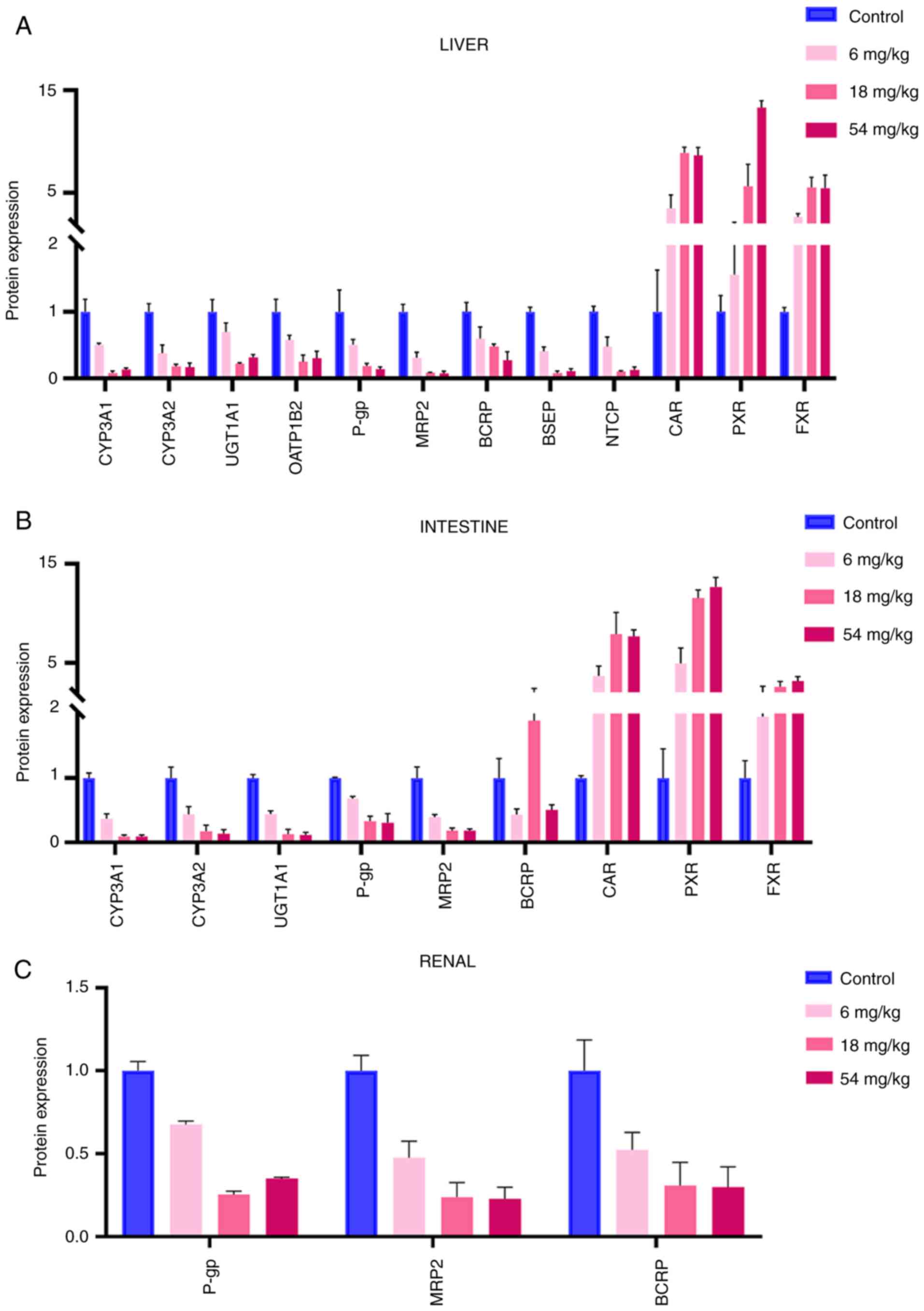 | Figure 4Effect of tripterine on protein
expression levels in various tissues. (A) Effect of tripterine on
the protein expression levels of CYP3A1, CYP3A2, UGT1A1, OATP1B2,
P-gp, BCRP, MRP2, BSEP, NTCP, CAR, PXR and FXR in the liver. (B)
Effect of tripterine on the protein expression levels of CYP3A1,
CYP3A2, UGT1A1, P-gp, BCRP, MRP2, CAR, PXR and FXR in the
intestine. (C) Effect of tripterine on the protein expression
levels of P-gp, BCRP and MRP2 in the renal tissues. The protein
expression levels in rats in the control and different doses of
tripterine groups were assessed by western blotting. Vertical bars
represent the mean ± SD (n=3). CYP, Cytochrome P450 proteins; UGT,
UDP-glucuronosyltransferases; OATP, organic anion transporting
polypeptides; P-gp, P-glycoprotein; BCRP, breast cancer resistance
protein; MRP, multi-drug resistance protein; BSEP, bile salt export
pump; NTCP, sodium taurocholate co-transporting polypeptide; CAR,
constitutive androstane receptor; PXR, pregnane X receptor; FXR,
farnesoid X receptor. |
 | Figure 5Western blotting images of
drug-metabolizing enzymes, drug transporters and nuclear receptors
in rat samples from each pre-treatment group. Representative
western blotting images in (A) liver, (B) in small intestine and
(C) in kidney. CYP, Cytochrome P450 proteins; UGT,
UDP-glucuronosyltransferases; OATP, organic anion transporting
polypeptides; P-gp, P-glycoprotein; BCRP, breast cancer resistance
protein; MRP, multi-drug resistance protein; BSEP, bile salt export
pump; NTCP, sodium taurocholate co-transporting polypeptide; CAR,
constitutive androstane receptor; PXR, pregnane X receptor; FXR,
farnesoid X receptor. |
Protein expression levels of DMEs, DTs
and NRs in the liver
As shown in Fig. 4A,
the protein expression of CYP3A1, CYP3A2, UGT1A1, OATP1B2, P-gp,
MRP2, BCRP, BSEP, NTCP, CAR, PXR and FXR in the liver was measured.
The protein expression of DMEs and DTs were found to be decreased,
whilst that of NRs was increased, compared with those in the
control group. The protein expression level of CYP3A1 in the three
tripterine pre-treated groups (6, 18 and 54 mg/kg) was decreased by
49.25, 91.47 and 86.88%, respectively. The decrease in the protein
expression level of CYP3A2 in the 6, 18 and 54 mg/kg tripterine
pre-treated groups was 29.26, 39.42 and 79.25%, respectively. The
protein expression level of UGT1A1 in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was decreased by 30.7, 77.83 and
68.07%, respectively. The protein expression level of OATP1B2 in
the 6, 18 and 54 mg/kg tripterine pre-treated groups was decreased
by 41.84, 74.97 and 69.8%, respectively. The reductions of the
protein expression level of P-gp in the 6, 18 and 54 mg/kg
tripterine pre-treated groups were by 49.64, 80.85 and 85.82%,
respectively. The protein expression level of BCRP in the 6, 18 and
54 mg/kg tripterine pre-treated groups was decreased by 40, 52.18
and 72.5%, respectively. Regarding MRP2, the expression of its
protein was reduced by 68.43, 91.6 and 92.62% in the 6, 18 and 54
mg/kg tripterine pre-treated groups. BSEP protein expression was
decreased by 58.98, 92.1 and 88.9%, respectively, in the 6, 18 and
54 mg/kg tripterine pre-treated groups. The protein expression
level of NTCP in the 6, 18 and 54 mg/kg tripterine pre-treated
groups was decreased by 51.70, 89.50 and 86.83%, respectively. The
protein expression level of CAR in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was increased by 249.55, 790.28 and
764.19%, respectively. The protein expression level of PXR in the
6, 18 and 54 mg/kg tripterine pre-treated groups was raised by
57.28, 465.38 and 1,231.11%, respectively. The increase of FXR
protein expression in the 6, 18 and 54 mg/kg tripterine pre-treated
groups was 167.03, 454.8 and 449.81%, respectively.
Protein expression levels of DMEs, DTs
and NRs in the intestine
As revealed in Fig.
4B, the protein expression of CYP3A1, CYP3A2, UGT1A1, P-gp,
MRP2, BCRP, CAR, PXR and FXR in the intestine was measured. The
protein expression level of CYP3A1 in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was found to be decreased by 62.9,
90.53 and 90.34%, respectively. The decrease of the protein
expression level of CYP3A2 in the 6, 18 and 54 mg/kg tripterine
pre-treated groups was 56.07, 82.61 and 86.31%, respectively. The
protein expression level of UGT1A1 in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was decreased by 55.66, 87 and
88.26%, respectively. The reduction of the protein expression level
of P-gp in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
31.78, 66.81 and 69.24%, respectively. The protein expression level
of BCRP in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
decreased by 46.96, 76.92 and 73.24%, respectively. For MRP2, the
expression of protein was reduced by 60.64, 81.78 and 81.36% in the
6, 18 and 54 mg/kg tripterine pre-treated groups, respectively. The
protein expression level of CAR in the 6, 18 and 54 mg/kg
tripterine pre-treated groups was increased by 268.82, 688.72 and
665.55%, respectively. The protein expression level of PXR in the
6, 18 and 54 mg/kg tripterine pre-treated groups was elevated by
397.40, 1,055.17 and 1,167.56%, respectively. The increase in FXR
protein expression in the 6, 18 and 54 mg/kg tripterine pre-treated
groups was 94.91, 158.49 and 215.79%, respectively.
Protein expression levels of DTs in
the kidney
As demonstrated in Fig.
4C, the protein expression of P-gp, MRP2 and BCRP in the kidney
tissues was measured. The protein expression level of P-gp in the
6, 18 and 54 mg/kg tripterine pre-treated groups was decreased by
32.47, 74.65 and 64.99%, respectively. The protein expression level
of BCRP in the 6, 18 and 54 mg/kg tripterine pre-treated groups was
decreased by 47.61, 69.27 and 70.29%, respectively. The protein
expression level of MRP2 in the 6, 18 and 54 mg/kg tripterine
pre-treated groups was decreased by 52.41, 76.12 and 77.17%,
respectively.
Discussion
Tripterygium wilfordii is a traditional
Chinese medicine with a wide range of pharmacological activities
that has been garnering significant attention over the past two
decades (8,32,40).
By contrast, CsA is one of the most commonly used long-term
immunosuppressive agents in clinical practice and has been shown in
numerous animal studies to have enhanced immunosuppressive effects
when combined with Tripterygium wilfordii after
transplantation whilst reducing its toxic effects. However, as a
natural herb, Tripterygium wilfordii contains a complex
mixture of compounds, making it challenging to elucidate its
underlying mechanisms precisely. To address this, tripterine, one
of the primary active components of Tripterygium wilfordii,
was focused upon to investigate whether it could regulate the
pharmacokinetics of CsA. To the best of our knowledge, the present
study was the first to demonstrate the dose-dependent inhibitory
effect of tripterine on the pharmacokinetics of CsA in rats. These
results suggest that pre-treatment with tripterine can
significantly reduce the bioavailability of CsA in rats.
In the present study, two important phase-I
metabolic enzymes, CYP3A1 and CYP3A2 were selected, along with the
widely distributed phase-II metabolic enzyme UGT1A1 as the targets
of investigation. CYP3A1 and CYP3A2 are primarily expressed in the
liver and small intestinal epithelial cells of rats, where they
metabolize ~70% CsA. Hou et al (41) previously reported that the
bioavailability of CsA was decreased when P-gp and CYP3A4 were
activated by glycyrrhizin. Based on this finding, it was
hypothesized that inhibiting CYP3A enzymes would likely increase
the bioavailability of CsA (36).
However, the present results showed that the expression of both
CYP3A1 and CYP3A2 was reduced, where the blood concentration of CsA
was also decreased, following tripterine administration. This
suggests that, in addition to its effects on drug-metabolizing
enzymes and transporters, tripterine likely regulated the
pharmacokinetics of CsA through other mechanisms.
OATP1B2 is typically expressed on the hepatocyte
sinusoidal membrane, with human homologs OATP1B1 and OATP1B3 being
closely related. They serve a role in the transport of linear and
cyclic peptides to the liver from the basal side of rat hepatocytes
(42,43). Both OATP1B2 and NTCP are influx
transporters that transport specific molecules from the blood into
the hepatic cytosol and are predominantly found on the basolateral
membrane of hepatocytes (44). By
contrast, P-gp, MRP2, BCRP and BSEP are efflux transporters located
on the canalicular membrane of hepatocytes. These transporters
facilitate the excretion of drugs and metabolites from the hepatic
cytosol into bile (44). In the
intestine, P-gp, MRP2 and BCRP, located on the apical side of
enterocytes, can pump drugs back into the gut lumen, thereby
limiting the oral absorption of CsA (45). In the kidney, P-gp, MRP2 and BCRP
are found on the basolateral side of proximal tubular cells, where
they pump drugs and their metabolites into the urine (46). CsA has been previously recognized to
be a broad-spectrum modulator for multi-drug resistance proteins,
such as P-gp, BCRP, MRP2 and OATP1B2 (47-50).
CAR, PXR and FXR are nuclear receptors that can regulate the
expression of various DMEs and DTs in a negative feedback manner,
including CYP3As, MRP2 and P-gp (51). The present study identified that
tripterine inhibited the expression of both DMEs (CYP3A1 and
CYP3A2) and DTs (OATP1B2, P-gp, BCRP and MRP2), likely by
activating CAR, PXR and FXR. However, whilst inhibition of P-gp and
BCRP would typically be expected to increase the plasma
concentration and decrease the clearance of their substrates (such
as CsA), results from the present study suggest a more complex
interaction at play.
In addition to the effects on DMEs and DTs, bile
acid metabolism serves a crucial role in the pharmacokinetics of
CsA. Previous studies have shown that the concentration of CsA
significantly decreases after bile duct ligation, suggesting the
importance of bile acids in its absorption and metabolism (52,53).
The present study demonstrated that tripterine had a profound
effect on bile-related transporters. Specifically, tripterine
significantly inhibited the expression of MRP2, NTCP and BSEP. NTCP
primarily mediates the uptake of bile salts into hepatocytes,
whilst BSEP and MRP2 are responsible for secreting mono-anionic and
di-anionic conjugated bile salts into bile (54,55).
By inhibiting the expression of NTCP, BSEP and MRP2 in the liver,
tripterine appeared to disrupt bile acid circulation. Furthermore,
tripterine was previously found to upregulate FXR expression in the
intestine, inhibiting bile acid synthesis through the FXR/FGF19
pathway (56). This suggests that
tripterine cannot only reduce bile acid synthesis but also regulate
bile acid metabolism. Given that the oral absorption of CsA is
known to depend on bile secretion (8,26,57),
the emulsification of bile acids aids in the absorption of CsA, a
macromolecular lipid-soluble drug, in the intestine (58). Therefore, findings from the present
study suggest that tripterine can decrease the oral bioavailability
of CsA by inhibiting bile acid metabolism and disrupting the normal
bile acid circulation process.
However, in the present study, only whole blood drug
concentration was detected to explore the pharmacokinetics of CsA,
but its intestinal and kidney drug concentration was not detected,
which is a limitation of the present study. The main components of
Tripterygium glycosides include Tripterygium triptolide, tripterine
and triptolide ketone. Different from Wuzhi capsule, which exerts
immunosuppressive effects by increasing the blood concentration of
CsA, multiple active components of Tripterygium glycosides possess
immunosuppressive properties, although its underlying mechanisms
remain elusive (59). Therefore, it
is highly likely that tripterine and CsA can exert
immunosuppression through different mechanisms, whereby the change
in plasma CsA concentration is not the only indicator that can be
used to evaluate their immune synergistic effects. The blood
concentration of CsA is not only closely associated with its
immunosuppressive efficacy but also to its toxicity. The primary
objective of the present study was to evaluate whether the
combination of tripterine and CsA can alter the pharmacokinetic
profile of the latter and to explore the underlying mechanisms, if
any. Associated pharmacodynamic or toxicity studies will be
required in subsequent studies. At present, the specific mechanism
of the association between the efficacy of CsA and its blood
concentration is lacking. Through the correlation analysis of
population pharmacokinetics, blood concentration was found to be
the dependent variable that had a positive correlation with the
efficacy of CsA. Further research will be needed to discover novel
easy-to-detect biochemical markers that can more specifically
represent the efficacy of CsA.
In conclusion, tripterine was found to inhibit the
expression of both DMEs and DTs, in addition to bile acid-related
transporters, which profoundly impacts bile acid circulation and
gastrointestinal lipid absorption. Given the significant positive
correlation between CsA absorption and lipid absorption, the
primary mechanism through which tripterine can reduce the oral
bioavailability of CsA appears to be its interference with lipid
absorption and bile acid metabolism. This inhibition disrupts the
normal absorption process of CsA, leading to a decrease in its
bioavailability. In addition, tripterine can dose-dependently
inhibit the oral bioavailability of CsA, which may be associated
with inhibition of the expression of bile acid transporters in
liver, activation of FXR in the intestine and the regulation of
bile acid metabolism.
Supplementary Material
Parent and product ions of the
analytes with individually optimized detection parameters.
Sequences of primers used for
PCR.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by National Natural
Science Foundation of China (grant nos. 82474008, 82173902 and
82104274).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SS, YL and RZ participated in research design and
supervision. PG and PL conducted experiments and confirm the
authenticity of all the raw data. JZ finished data analysis and
visualization of data. JZ and RZ wrote or contributed to the
writing of the manuscript. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
All animal studies were approved (approval no. 4352;
date, 2023) by The Institutional Animal Care and Use Committee of
Tongji Medical College Huazhong University of Science and
Technology (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lv H, Jiang L, Zhu M, Li Y, Luo M, Jiang
P, Tong S, Zhang H and Yan J: The genus Tripterygium: A
phytochemistry and pharmacological review. Fitoterapia.
137(104190)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Liu L, Luo Y, Zhou M, Lu Y, Xing M, Ru Y,
Sun X, Chen X, Li S, Hong S, et al: Tripterygium agents for the
treatment of atopic eczema: A Bayesian analysis of randomized
controlled trials. Phytomedicine. 59(152914)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang D, Zhang H, Liang J, Wang H, Hua B,
Feng X, Gilkeson GS, Farge D, Shi S and Sun L: A long-term
follow-up study of allogeneic mesenchymal stem/stromal cell
transplantation in patients with drug-resistant systemic lupus
erythematosus. Stem Cell Reports. 10:933–941. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lü S, Wang Q, Li G, Sun S, Guo Y and Kuang
H: The treatment of rheumatoid arthritis using Chinese medicinal
plants: From pharmacology to potential molecular mechanisms. J
Ethnopharmacol. 176:177–206. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ramgolam V, Ang SG, Lai YH, Loh CS and Yap
HK: Traditional Chinese medicines as immunosuppressive agents. Ann
Acad Med Singap. 29:11–16. 2000.PubMed/NCBI
|
|
6
|
Jiang X, Huang XC, Ao L, Liu WB, Han F,
Cao J, Zhang DY, Huang CS and Liu JY: Total alkaloids of
Tripterygium hypoglaucum (levl.) Hutch inhibits tumor growth both
in vitro and in vivo. J Ethnopharmacol. 151:292–298.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang YQ, Wu YF, Xu FF, Deng JB, Wu LL, Han
XD, Liang J, Guo DA and Liu B: Tripterygium glycoside fraction n2:
Alleviation of DSS-induced colitis by modulating immune homeostasis
in mice. Phytomedicine. 58(152855)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li JM, Jiang Q, Tang XP, Yang H and Zhou
ZQ: Study advances in regulation effect of Tripterygium
wilfordii and its extracts on innate immune system in
rheumatoid arthritis cases. Zhongguo Zhong Yao Za Zhi.
44:3384–3390. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
9
|
Liu L, Yan J, Shu JC and Liu JQ: Advance
on alkaloids from Tripterygium wilfordii and their
bioactivities. Nat Prod Res Dev. 31:2170–2181. 2019.
|
|
10
|
Lipsky PE and Tao XL: A potential new
treatment for rheumatoid arthritis: Thunder god vine. Semin
Arthritis Rheum. 26:713–723. 1997.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Brinker AM, Ma J, Lipsky PE and Raskin I:
Medicinal chemistry and pharmacology of genus Tripterygium
(Celastraceae). Phytochemistry. 68:732–766. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nong C, Wang XZ, Jiang ZZ and Zhang LY:
Progress of effect and mechanisms of Tripterygium wilfordii
on immune system. Zhongguo Zhong Yao Za Zhi. 44:3374–3383.
2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
13
|
Wu X, Huang W, Guo B, Si J and Zhang J:
Determination of contents of tripterine in Tripterygii
preparations. Zhongguo Zhong Yao Za Zhi. 34:836–838.
2009.PubMed/NCBI(In Chinese).
|
|
14
|
Li H, Fan Y, Yang F, Zhao L and Cao B: The
coordinated effects of Apatinib and Tripterine on the
proliferation, invasiveness and apoptosis of human hepatoma Hep3B
cells. Oncol Lett. 16:353–361. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen YW, Lin GJ, Hueng DY, Huang SH, Chia
WT, Shieh YS, Ma KH and Sytwu HK: Enhanced anti-tumor activity of
triptolide in combination with irradiation for the treatment of
oral cancer. Planta Med. 80:255–261. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xiong Y, Yan Y and Li Y: RETRACTED:
Tripterine alleviates LPS-induced inflammatory injury by
up-regulation of miR-146a in HaCaT cells. Biomed Pharmacother.
105:798–804. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li X, Lu Q, Xie W, Wang Y and Wang G:
Anti-tumor effects of triptolide on angiogenesis and cell apoptosis
in osteosarcoma cells by inducing autophagy via repressing
Wnt/β-catenin signaling. Biochem Biophys Res Commun. 496:443–449.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen Y, Qu D, Fu R, Guo M, Qin Y, Guo J
and Chen Y: A Tf-modified tripterine-loaded coix seed oil
microemulsion enhances anti-cervical cancer treatment. Int J
Nanomedicine. 13:7275–7287. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zuo A, Zhao P, Zheng Y, Hua H and Wang X:
Tripterine inhibits proliferation, migration and invasion of breast
cancer MDA-MB-231 cells by up-regulating microRNA-15a. Biol Chem.
400:1069–1078. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yuan L, Liu C, Chen Y, Zhang Z, Zhou L and
Qu D: Antitumor activity of tripterine via cell-penetrating
peptide-coated nanostructured lipid carriers in a prostate cancer
model. Int J Nanomedicine. 8:4339–4350. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jin C, He X, Zhang F, He L, Chen J, Wang
L, An L and Fan Y: Inhibitory mechanisms of celastrol on human
liver cytochrome P450 1A2, 2C19, 2D6, 2E1 and 3A4. Xenobiotica.
45:571–577. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sun M, Tang Y, Ding T, Liu M and Wang X:
Inhibitory effects of celastrol on rat liver cytochrome P450 1A2,
2C11, 2D6, 2E1 and 3A2 activity. Fitoterapia. 92:1–8.
2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang YS, Tu YY, Gao XC, Yuan J, Li G,
Wang L, Deng JP, Wang Q and Ma RM: Strong inhibition of celastrol
towards UDP-glucuronosyl transferase (UGT) 1A6 and 2B7 indicating
potential risk of UGT-based herb-drug interaction. Molecules.
17:6832–6839. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhao Q, Liu F, Cheng Y, Xiao XR, Hu DD,
Tang YM, Bao WM, Yang JH, Jiang T, Hu JP, et al: Celastrol protects
from cholestatic liver injury through modulation of SIRT1-FXR
signaling. Mol Cell Proteomics. 18:520–533. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hua H, Zhang Y, Zhao F, Chen K, Wu T, Liu
Q, Huang S, Zhang A and Jia Z: Celastrol inhibits intestinal lipid
absorption by reprofiling the gut microbiota to attenuate high-fat
diet-induced obesity. iScience. 24(102077)2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lindholm A, Henricsson S and Dahlqvist R:
The effect of food and bile acid administration on the relative
bioavailability of cyclosporin. Br J Clin Pharmacol. 29:541–548.
1990.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gupta SK, Manfro RC, Tomlanovich SJ,
Gambertoglio JG, Garovoy MR and Benet LZ: Effect of food on the
pharmacokinetics of cyclosporine in healthy subjects following oral
and intravenous administration. J Clin Pharmacol. 30:643–653.
1990.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yang X, Xu L, Ren H, Li Z, Sun C and Zhang
Z: The effect of multi-glycosides of Tripterygium wilfordii
Hook f. (GTW) on the survival time of cardiac allografts in rats.
Chin Med Sci J. 7:232–234. 1992.PubMed/NCBI
|
|
29
|
Zhang LL and Sun JH: Synergistic effects
of Tripterygium wilfordii polyglycosides and cyclosporine A
in rat heart transplantation. Chin J Exp Surg. 23–24. 2001.
|
|
30
|
Li J and Hao J: Treatment of
neurodegenerative diseases with bioactive components of
Tripterygium wilfordii. Am J Chin Med. 47:769–785.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang HL, Jiang Q, Feng XH, Zhang HD, Ge L,
Luo CG, Gong X and Li B: Tripterygium wilfordii Hook F
versus conventional synthetic disease-modifying anti-rheumatic
drugs as monotherapy for rheumatoid arthritis: A systematic review
and network meta-analysis. BMC Complement Altern Med.
16(215)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang XB, Dai EL, Xue GZ and Ma RL: A
PRISMA-compliant systematic review and network meta-analysis on the
efficacy between different regimens based on Tripterygium
wilfordii Hook F in patients with primary nephrotic syndrome.
Medicine (Baltimore). 97(e11282)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yu CP, Lin HJ, Lin SP, Shia CS, Chang PH,
Hou YC and Hsieh YW: Rhubarb decreased the systemic exposure of
cyclosporine, a probe substrate of P-glycoprotein and CYP 3A.
Xenobiotica. 46:677–682. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dupuis R, Yuen A and Innocenti F: The
influence of UGT polymorphisms as biomarkers in solid organ
transplantation. Clin Chim Acta. 413:1318–1325. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yigitaslan S, Erol K and Cengelli C: The
effect of P-glycoprotein inhibition and activation on the
absorption and serum levels of cyclosporine and tacrolimus in rats.
Adv Clin Exp Med. 25:237–242. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Goldberg H, Ling V, Wong PY and Skorecki
K: Reduced cyclosporin accumulation in multidrug-resistant cells.
Biochem Biophys Res Commun. 152:552–558. 1988.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Fahr A: Cyclosporin clinical
pharmacokinetics. Clin Pharmacokinet. 24:472–495. 1993.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Huang X, Zhang R, Yang T, Wei Y, Yang C,
Zhou J, Liu Y and Shi S: Inhibition effect of
epigallocatechin-3-gallate on the pharmacokinetics of calcineurin
inhibitors, tacrolimus, and cyclosporine A, in rats. Expert Opin
Drug Metab Toxicol. 17:121–134. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang T, Liu Y, Huang X, Zhang R, Yang C,
Zhou J, Zhang Y, Wan J and Shi S: Quercetin-3-O-β-D-glucoside
decreases the bioavailability of cyclosporin A through regulation
of drug metabolizing enzymes, transporters and nuclear receptors in
rats. Mol Med Rep. 18:2599–2612. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29(e45)2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hou YC, Lin SP and Chao PDL: Liquorice
reduced cyclosporine bioavailability by activating P-glycoprotein
and CYP 3A. Food Chem. 135:2307–2312. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Manikandan P and Nagini S: Cytochrome P450
structure, function and clinical significance: A review. Curr Drug
Targets. 19:38–54. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Meier-Abt F, Faulstich H and Hagenbuch B:
Identification of phalloidin uptake systems of rat and human liver.
Biochim Biophys Acta. 1664:64–69. 2004.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Pan G: Roles of hepatic drug transporters
in drug disposition and liver toxicity. Adv Exp Med Biol.
1141:293–340. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Müller J, Keiser M, Drozdzik M and Oswald
S: Expression, regulation and function of intestinal drug
transporters: An update. Biol Chem. 398:175–192. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ivanyuk A, Livio F, Biollaz J and Buclin
T: Renal drug transporters and drug interactions. Clin
Pharmacokinet. 56:825–892. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Li L, Yao QQ, Xu SY, Hu HH, Shen Q, Tian
Y, Pan LY, Zhou H, Jiang HD, Lu C, et al: Cyclosporin A affects the
bioavailability of ginkgolic acids via inhibition of P-gp and BCRP.
Eur J Pharm Biopharm. 88:759–767. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Xia CQ, Liu N, Miwa GT and Gan LS:
Interactions of cyclosporin a with breast cancer resistance
protein. Drug Metab Dispos. 35:576–582. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Akashi M, Tanaka A and Takikawa H: Effect
of cyclosporin A on the biliary excretion of cholephilic compounds
in rats. Hepatol Res. 34:193–198. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Tang W, Stearns RA, Chen Q, Bleasby K,
Teffera Y, Colletti A, Hafey M, Evers R, Dean DC, Magriotis PA, et
al: Importance of mechanistic drug metabolism studies in support of
drug discovery: A case study with an N-sulfonylated dipeptide VLA-4
antagonist in rats. Xenobiotica. 38:223–237. 2008.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Xu C, Li CYT and Kong ANT: Induction of
phase I, II and III drug metabolism/transport by xenobiotics. Arch
Pharm Res. 28:249–268. 2005.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Venkataramanan R, Perez HD, Schwinghammer
T, Burckart GJ, Ptachcinski RJ, Van Thiel DH and Starzl TE: Effect
of bile on cyclosporine absorption in dogs. Res Commun Chem Pathol
Pharmacol. 53:137–140. 1986.PubMed/NCBI
|
|
53
|
Takaya S, Iwatsuki S, Noguchi T, Koie H,
Zaghloul I, Venkataramanan R and Starzl TE: The influence of liver
dysfunction on cyclosporine pharmacokinetics-a comparison between
70 per cent hepatectomy and complete bile duct ligation in dogs.
Jpn J Surg. 19:49–56. 1989.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Meier PJ and Stieger B: Bile salt
transporters. Annu Rev Physiol. 64:635–661. 2002.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Stieger B: The role of the
sodium-taurocholate cotransporting polypeptide (NTCP) and of the
bile salt export pump (BSEP) in physiology and pathophysiology of
bile formation. Handb Exp Pharmacol. 205–259. 2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Katafuchi T and Makishima M: Molecular
basis of bile acid-FXR-FGF15/19 signaling axis. Int J Mol Sci.
23(6046)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Lindholm A: Factors influencing the
pharmacokinetics of cyclosporine in man. Ther Drug Monit.
13:465–477. 1991.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Guan P, Lu Y, Qi J, Niu M, Lian R, Hu F
and Wu W: Enhanced oral bioavailability of cyclosporine A by
liposomes containing a bile salt. Int J Nanomedicine. 6:965–974.
2011.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Fan J, Chen L, Lu X, Li M and Zhu L: The
pharmacokinetic prediction of cyclosporin A after coadministration
with Wuzhi capsule. AAPS PharmSciTech. 20(247)2019.PubMed/NCBI View Article : Google Scholar
|