Introduction
Tricho-hepato-enteric (THES) syndrome is a rare and
severe congenital diarrheal disorder, also known as syndromic
diarrhea (1). With a prevalence of
~1 in 1,000,000 births, only 96 neonatal cases have been reported
to date (2). Currently, there is
only one single incidental reference regarding its prenatal
diagnosis (3). The present case
report seeks to expand the knowledge base by offering novel
insights into the prenatal ultrasound findings associated with THES
syndrome.
Historically, Stankler et al (4) first described this condition in 1982,
followed in 1994 by Girault et al (5) THES is classified into two types, THES1
and THES2, which are caused by homozygous or compound heterozygous
mutations in the SKI3 subunit of superkiller complex (SKIC3,
formerly TTC37) and SKI2 subunit of superkiller complex
(SKIC2, formerly SKIV2L) genes, respectively
(6). Intractable diarrhea (100% of
cases) that persists even with bowel rest is the hallmark symptom
and typically manifests within the first days of life but can be
delayed up to 8 months (2). Other
phenotypic findings include hair abnormalities (90%) such as thin,
sparse, wooly (trichorrexis nodosa) and brittle hair, significant
facial abnormalities (84%) such as a prominent forehead and cheeks,
broad nose, long philtrum and hypertelorism. IUGR/SGA is frequent,
observed in 70% of the SKIC3-mutated and 86% of the
SKIC2-mutated patients clinically explored (2). Most affected children fall below the
10th percentile (30 cases) in growth charts or even below the 3rd
centile (27 cases) (1). A defective
immune system is frequently diagnosed (50%) with low immunoglobulin
levels and platelet disorders are present in 73% of cases (2). Skin abnormalities, such as
‘café-au-lait’ spots, are observed in 39-53% of patients (2). Liver disorders, including cholestatic
jaundice, cirrhosis, or siderosis, are more prevalent in
SKIC2-mutated patients (88%) compared with those with
SKIC3-mutations (51%) (2).
Rare cardiac defects (20%), such as aortic insufficiency, septal
defects, peripheral pulmonary stenosis, and Tetralogy of Fallot,
have also been documented (1,2).
THES syndrome is part of a broader family of
congenital diarrheal disorders, which also includes microvillous
inclusion disease (MVID) and congenital tufting enteropathy
(7). These conditions share the
life-threatening intractable diarrhea requiring parenteral
nutrition and intestinal transplantation (6,7).
Despite differing histological and genetic profiles, their
pathogenic mechanisms appear similar. Specifically, intestinal
biopsies from patients with THES revealed mild to severe villous
atrophy in the intestinal brush border (6,8). The
primary function of the microvillous brush border is the formation
of a surface responsible for absorption and digestion. Notably,
villi begin to develop in the fetus around the ninth week of
gestation (9).
Prenatal diagnosis of THES remains limited due to
its rarity. By contrast, conditions such as MVID frequently present
with detectable prenatal ultrasonographic abnormalities,
particularly in the third trimester. According to Sun et al
(10), among 47 cases of MVID,
ultrasound findings revealed echogenic bowel, bowel dilation, and
polyhydramnios. The sole prenatal case of the THES was incidentally
reported by Zhou et al (3),
involving a patient with a homozygous mutation in SKIC2. In
the present case, additional ultrasonographic findings were
included, and the prenatal association will be further
explored.
Case presentation
The present study was a case report of a 34-year-old
healthy pregnant woman G2P2 who was referred for a mid-trimester
fetal anomaly ultrasound scan due to fetal growth retardation of
the fetus. The pregnancy and delivery of the first child was
uneventful. The couple did not report consanguinity and the family
history was not significant. The patient denied any teratogenic
exposure or bleeding during the pregnancy. The gestational age was
22 weeks and 4 days based on the crown rump length of the
first-trimester scan. The measurements of detailed fetal biometry
were performed and noted below. The INTERGROWTH-21st charts and
their measurement techniques for all parameters were used.
Biparietal diameter (BPD), head circumference (HC),
abdominal circumference (AC) and Estimated Fetal Weight-fell below
the 3rd centile for the gestational age. The only exception was the
occipital-frontal diameter (OFD), evaluated at the 43.9th centile.
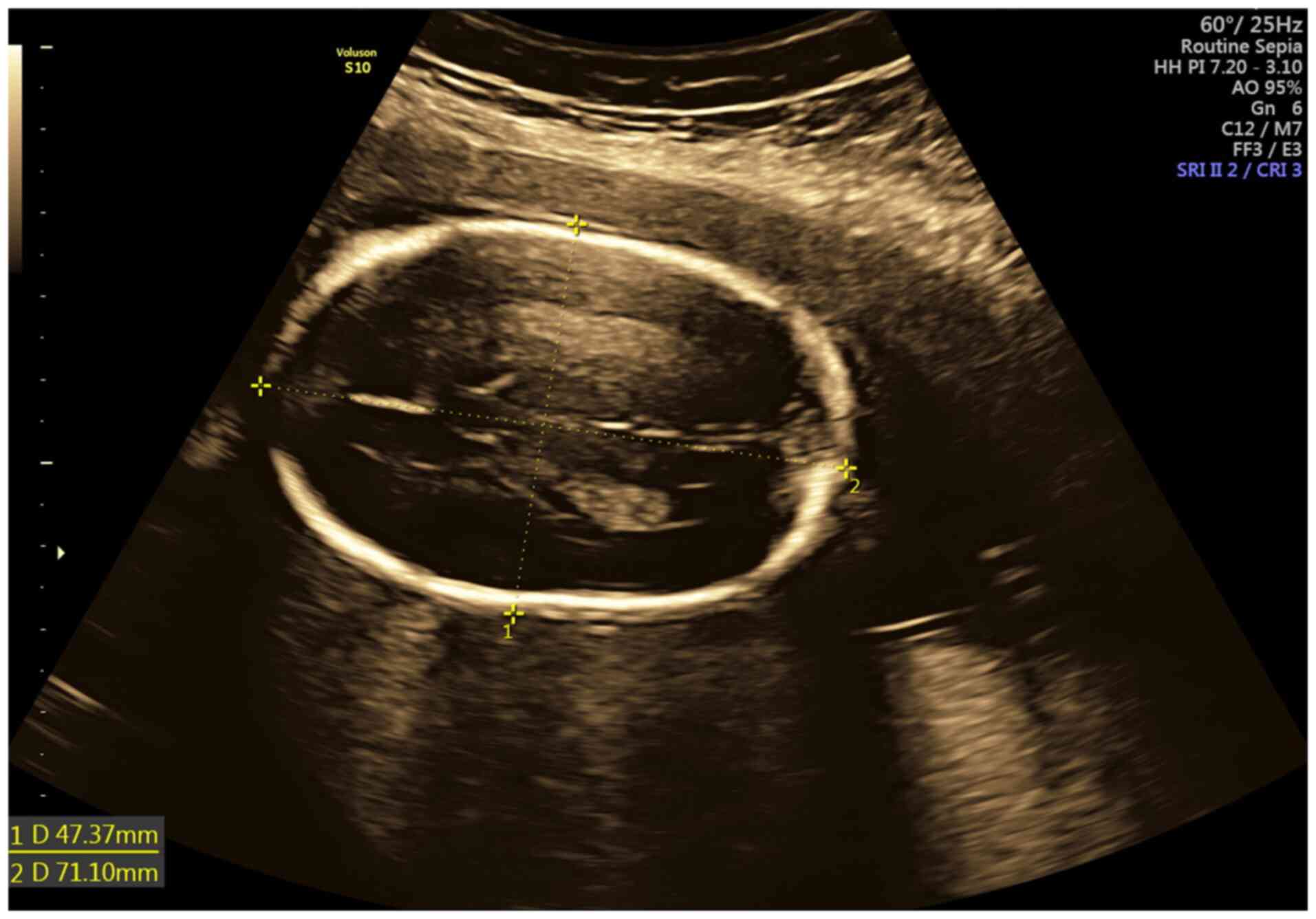
The ratio BPD/OFD corresponded to the cephalic index of 0.67 (CI
normal range: 0.74-0.83) which is indicative of dolichocephaly
(Fig. 1). Concerning the long
bones, both extremities were underdeveloped below the 3rd centile.
No structural anomalies were detected in the fetal organs, the
fetal sex was male, and the amniotic fluid was normal by the
measurement of the deepest pocket. Doppler parameters for the
median uterine artery (PI 25.7th percentile) and umbilical artery
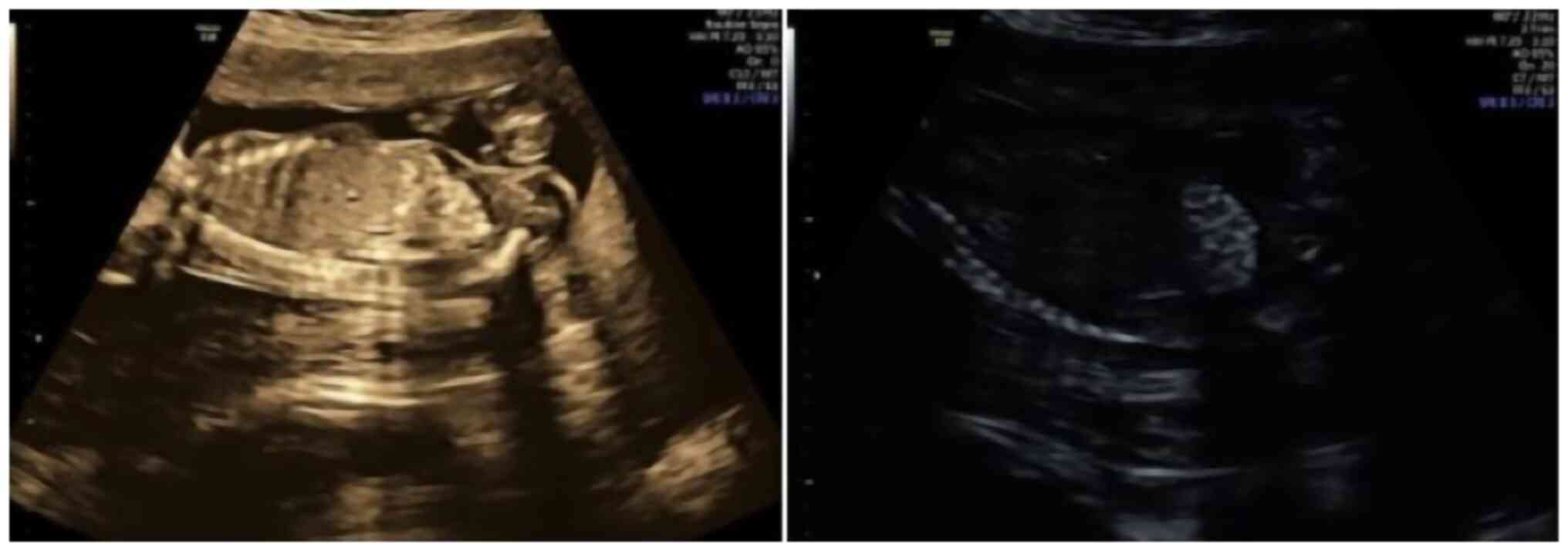
(PI 37.8th percentile) were within normal limits. The bowel
appeared echogenic according to the criteria developed by Slotnick
and Abuhamad (11), with a grade 3
based on echogenicity relative to the surrounding bone, such as the
fetal iliac crest (12) (Fig. 2).
Based on the International Society of Ultrasound in
Obstetrics and Gynecology guidelines (13), the diagnosis indicates an early
onset of apparently isolated fetal growth restriction (FGR)
accompanied by dolichocephaly and a severe form of echogenic bowel
(EB). Given these findings, the authors proceeded with an
amniocentesis to obtain a sample of amniotic fluid for the purpose
of prenatal testing and a molecular analysis to determine the
underlying causes of the FGR.
Due to the superior diagnostic yield of Whole Exome
Sequencing (WES), which ranges from 4-12% in cases of isolated FGR
(14,15), compared with chromosomal microarray
analysis (CMA) (16,17) it was decided to proceed with WES.
Additionally, WES was performed on both parents, implementing a
Trio-WES approach. DNA was isolated from the amniotic fluid sample
and parental peripheral blood. Following exome amplification,
sequencing by next generation sequencing was performed using the
NovaSeq 600 Sequencing System (Illumina, Inc.). The samples were
prepared for sequencing using Nonacus Cell3 Target Nexome (cat. no.
NGS_C3T_NEX_FR; Nonacus) and NovaSeq 6000 SP Reagent Kit v1.5 (300
cycles) (cat. no. 20028400). The final library concentration was
calculated by Qubit dsDNA BR assay kit (Invitrogen; Thermo Fisher
Scientific, Inc.) and 0,9 nM of loading concentration was used.
Short Reads Pair End sequencing of 150 bp was performed using the
NovaSeq 6000 Sequencing System (Illumina, Inc.). Quality and
integrity of processed samples was assessed using the log file
generated by DRAGEN, which provides the exact depth and metadata of
the index case. Demultiplexing of the sequencing data was performed
with Binary Base Call BCL Convert (Illumina, Inc.) and Alignment
and Variant Calling with DRAGEN Bio-IT platform (Illumina, Inc.).
Fastq files were uploaded and all variants in genes, associated
with known genetic disorders and syndromes (according to the OMIM
database), were analyzed using Varsome Clinical (Saphetor) and the
bioinformatics databases.
All genes that are associated with known genetic
disorders and syndromes (according to the OMIM database) were
analyzed using Varsome Clinical (Saphetor) and other bioinformatic
tools, including Splice AI (https://spliceailookup.broadinstitute.org/), Primate
AI (https://github.com/Illumina/PrimateAI), CADD
(https://cadd.gs.washington.edu/), REVEL
(https://sites.google.com/site/revelgenomics/) and SIFT
(https://sift.bii.a-star.edu.sg/).
All findings were evaluated according to the
international literature and the American College of Medical
Genetics and Genomics (ACMG) guidelines (18). The reference genome was
GRCh37/hg19.
Bioinformatic analysis of the Trio-WES results
revealed that the embryo was compound heterozygous for two nonsense
mutations in the SKIC2 gene. The embryo carried the
c.3187C>T (p. Arg1063Ter) and the c.1528C>T (p. Arg510Ter)
mutations in SKIC2, inherited from its mother and father,
respectively. Mutation c.3187C>T (Variation ID: 1323596) causes
a premature stop codon in exon 26 (of 28) while mutation
c.1528C>T (Variation ID: 2152010) causes a premature stop codon
in exon 14. Both mutations are expected to disrupt the form and
function of the produced protein and are categorized as pathogenic
based on the ACMG/AMP guidelines (18).
Moreover, bioinformatic analysis of the Trio-WES
results revealed that the embryo was also compound heterozygous for
two missense mutations in the SERPINA1 gene. The embryo
carried the c.1177C>T (p.Pro393Ser) and the c.839A>T
(p.Asp280Val) mutations in SERPINA1, inherited from its father and
mother, respectively. Both c.1177C>T (Variation ID: 289135) and
c.839A>T (Variation ID: 17975) are categorized as pathogenic
based on the ACMG/AMP guidelines (18).
Simultaneously, quantitative fluorescence (QF) PCR,
conventional karyotype, and CMA (a-CGH) were performed to reduce
turnaround time. An extensive molecular analysis of the CF
transmembrane regulator (CFTR) gene to establish the
CFTR mutation spectrum was also performed. The results were
normal.
While awaiting the results, a follow-up ultrasound
was performed at 28 weeks and 4 days' gestation. The biometric
measurements, including BPD, AC, and all long bone lengths,
remained below the 3rd percentile, indicating no significant
changes. However, the OFD had increased to the 91.8th percentile,
leading to a lower cephalic index of 0.65. The amniotic fluid index
was found to be within normal limits. It has been reconfirmed that
there are no structural anomalies. Furthermore, Doppler assessments
for the median uterine artery (PI at the 24.9th percentile) and
umbilical artery (PI at the 65.8th percentile) were also
normal.
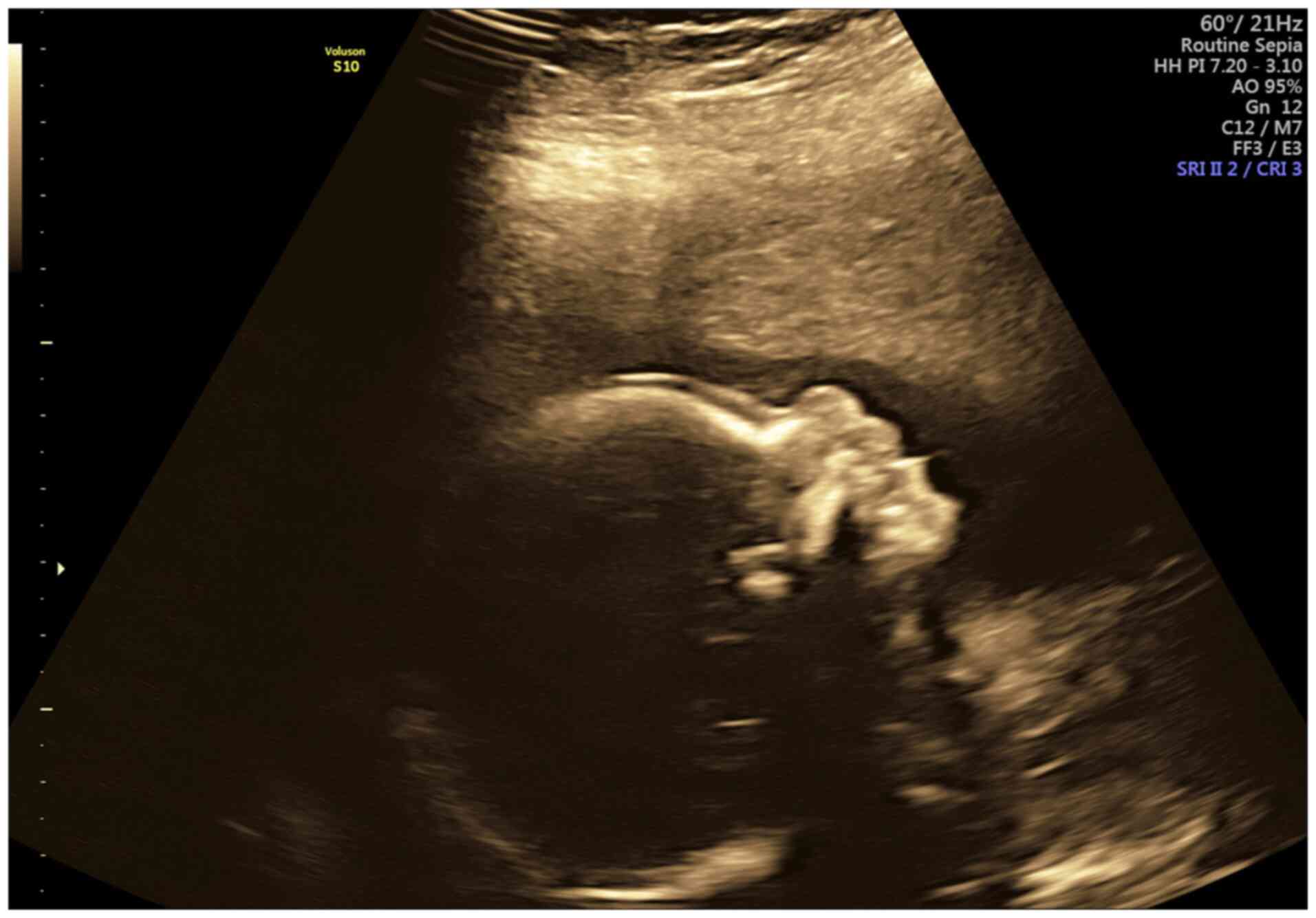
In retrospect, following the diagnosis of THES and
given the well-known clinical feature of a prominent forehead, the
facial angle was measured and found to be within the normal range
(128.12˚±10.99˚) (19) (Fig. 3).
The patient was counseled regarding the anticipated
spectrum of neonatal outcomes. After considering the information,
the couple opted for the termination of the pregnancy. Fetal demise
was induced via an intra-amniotic injection of Digoxin
administered 24 h before the scheduled abortion procedure.
Ultimately, the couple chose not to proceed with a postmortem
examination of the fetus.
Discussion
The FGR of the presented case has distinctive
characteristics that prompt consideration of various potential
etiologies, which can be included or excluded from the present
diagnostic work-up. Firstly, the FGR was notable for the absence of
any ‘ultrasound apparent structural anomalies’ except the EB (soft
marker). Secondly, the condition is not associated with abnormal
Doppler parameters, thereby allowing to exclude placental
insufficiency as a primary cause (20). Thirdly, it is manifested as an early
onset FGR which is symmetrical across all biometric parameters of
the fetus according to the gestational age.
Given that there were no other obvious risk factors
from maternal history (maternal age, exposure to toxins and
substance abuse) and negative results from the mother's serological
tests for TORCH infections, the focus must shift towards genetic
etiologies, including chromosomal anomalies, submicroscopic and
monogenic disorders (20).
Chromosomal anomalies contribute to 19% of fetal FGR
cases, with isolated chromosomal anomalies accounting for 3.4-9.6%.
Aneuploidy is the most common, in 5.8% of cases, with triploidy
predominant 26 weeks before and trisomy 18 more prevalent after.
Occasionally FGR in trisomy 21 is associated with placental
deficiency and abnormal umbilical artery impedance (20). Confined placental mosaicism,
particularly trisomy 16, occurs in 9-16% of cases (20,21).
Submicroscopic anomalies (for example, microdeletions/duplications)
account for 4% of isolated FGR cases (17), and monogenic disorders range from
4-12% (14,15), with equal postnatal prevalence at
11% (22). Meler et al
(20) classify FGR into two groups:
Symmetric FGR, affecting all fetal biometry and linked to syndromic
features the present case report and asymmetric FGR, with only
short long bones (below 3SD), often due to skeletal dysplasia.
The second significant finding from the fetal
anomaly scan was the presence of a severe EB, a condition observed
in ~0.2 to 1.8% of cases, which can occasionally be transient. A
systematic review by D'Amico et al (23) for the incidence of isolated EB
identified chromosomal anomalies (primarily trisomy 21), in 3.3%,
cystic fibrosis in 2.2%, congenital infections (mostly CMV) in
2.2%, and gastrointestinal anomalies in 2.1% of cases. EB coexists
with FGR in 12.6% of cases, The most likely mechanism involved the
redistribution of blood flow away from the splanchnic circulation
towards more vital organs such as the brain, resulting in
mesenteric ischemia or edema of the intestinal wall and
consequently in intestinal dysfunction. In the context of THES,
this hypothesis is doubtful due to the absence of any signs of
placental insufficiency or ischemia. Thus, villous atrophy remains
the only plausible explanation for the intestinal dysfunction
observed. Nevertheless, this hypothesis warrants further
confirmation. By the authors' diagnostic work-up, all known EB
etiologies were ruled out and a follow-up scan in 3rd trimester
revealed no gastrointestinal anomalies. EB has also been linked to
MVID (another form of congenital diarrhea), with a 7% incidence,
occasionally presenting with polyhydramnios but without postnatal
bowel obstruction (10).
The severe dolichocephaly of the fetus lacks any
discernible intrauterine cause, such as breech presentation or
oligohydramnios and any cranial deformity indicative of sagittal
craniosynostosis (24,25). This condition likely results in the
prominent postnatal forehead described in most cases in the
literature in the cases of THES (26). While dolichocephaly is
well-explained in cases of sagittal synostosis, its occurrence in
the absence of such synostosis according to a theory is attributed
to a modular growth driven by gene transcription between the
tissues, which can describe any level of organization, for example,
the calvarium or specific areas of the cranial vault (27).
The results of the Trio-WES that was performed
revealed that the embryo had compound heterozygous mutations in the
SKIC2 gene which it had inherited from its parents.
Homozygous and compound heterozygous mutations in SKIC2 have
been associated with THES2 (OMIM#614602). Trio-WES results also
revealed that the embryo was also compound heterozygous for
mutations in the SERPINA1 gene. Mutations in SERPINA1
have been associated with Alpha-1-antitrypsin (AAT) deficiency
(OMIM#613490). The most common symptom of AAT deficiency is
emphysema which appears in the 3rd-4th decade and a less common
symptom is liver disease which can appear in the neonatal period
(28). The SERPINA1 gene
mutations were not expected to have contributed to the fetal
phenotype.
From the literature, the only case of prenatal
diagnosis of THES is in a study of 51 fetuses with isolated and
severe FGR where Trio-WES was performed (3). One of them had a homozygous
SKIC2 mutation, causing THES. The only difference compared
with the present case is the asymmetrical pattern of FGR. The fetus
was preterm born at 36 weeks due to fetal distress.
The present study contributed valuable novel
prenatal findings on THES, enriching the limited existing
literature and building upon the few up-to-date studies in prenatal
settings. Moreover, the present study highlighted a significant
knowledge gap, emphasizing the need for further investigations.
Finally, it potentially provided insights into diagnostic
challenges faced by clinicians. In detail, it revealed the
significance of the use of Trio-WES as part of the diagnostic
work-up for FGR, particularly in cases without any pathognomic
findings. Therefore, it has the potential to influence clinical
guidelines, patient care, and future research.
The present case report has several limitations.
First, it presented a single case rather than a case series due to
the extreme rarity of THES. Secondly, it lacked histopathological
evidence, which could have provided additional valuable insights.
Furthermore, the absence of postnatal data and longitudinal
follow-up, although justified by the mother's personal choice for
termination-leads to an unknown clinical course of the disorder and
limits the predictive value of prenatal findings for patient
outcomes. Given that this is one of the first reports describing
this rare genetic disorder, the present study did not allow for an
investigation of causal relationships due to the lack of
comparative studies and controls.
Given the extreme rarity of this disorder, the
prenatal findings alone may not be immediately applicable to
real-world clinical practice. However, due to the valuable novel
insights provided by this case report, these findings should be
considered for further research and the broader application of
fetal sequencing, particularly whole-exome sequencing (WES), in
prenatal settings. Expanding such investigations will help identify
a larger number of cases, thereby enhancing the external validity
of the results.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the European Nucleotide Archive (ENA), under accession number
PRJEB85611 or at the following URL: https://www.ebi.ac.uk/ena/browser/text-search?query=PRJEB85611.
Authors' contributions
AlM, AnM and NZ substantially contributed to the
conception and the design of the study, contributed to manuscript
drafting or critical revisions on the intellectual content,
approved the final manuscript version to be published, agreed to be
accountable for all aspects of the work, so that any questions
relating to research integrity or scientific accuracy in any part
of the present study are appropriately investigated and resolved.
IP performed quantitative fluorescence PCR, conventional karyotype,
CMA (a-CGH) and bioinformatic analysis of the Trio-WES, contributed
to manuscript drafting or critical revisions on the intellectual
content, approved the final manuscript version to be published,
agreed to be accountable for all aspects of the work, so that any
questions relating to research integrity or scientific accuracy in
any part of the study are appropriately investigated and resolved.
AA agreed to be accountable for all aspects of the work and to have
a major role in patient consultation for the treatment plan, so
that any questions relating to research integrity or scientific
accuracy in any part of the study are appropriately investigated
and resolved. AlM, AnM, NZ and IP confirm the authenticity of all
the raw data. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The parents provided written informed consent for
the publication of their data (including their fetus) and the fetal
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fabre A, Martinez-Vinson C, Goulet O and
Badens C: Syndromic diarrhea/Tricho-hepato-enteric syndrome.
Orphanet J Rare Dis. 8(5)2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bourgeois P, Esteve C, Chaix C, Béroud C
and Lévy N: THES clinical consortium. Fabre A and Badens C:
Tricho-Hepato-Enteric Syndrome mutation update: Mutations spectrum
of TTC37 and SKIV2L, clinical analysis and future prospects. Hum
Mutat. 39:774–789. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhou H, Fu F, Wang Y, Li R, Li Y, Cheng K,
Huang R, Wang D, Yu Q, Lu Y, et al: Genetic causes of isolated and
severe fetal growth restriction in normal chromosomal microarray
analysis. Int J Gynaecol Obstet. 161:1004–1011. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Stankler L, Lloyd D, Pollitt RJ, Gray ES,
Thom H and Russell G: Unexplained diarrhea and failure to thrive in
2 siblings with unusual facies and abnormal scalp hair shafts: A
new syndrome. Arch Dis Child. 57:212–216. 1982.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Girault D, Goulet O, Le Deist F, Brousse
N, Colomb V, Césarini JP, de Potter S, Canioni D, Griscelli C,
Fischer A, et al: Intractable infant diarrhea associated with
phenotypic abnormalities and immunodeficiency. J Pediatr.
125:36–42. 1994.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fabre A, Bourgeois P, Chaix C, Bertaux K,
Goulet O and Badens C: Trichohepatoenteric Syndrome. In:
GeneReviews. Adam MP, Feldman J and Mirzaa GM (eds). University of
Washington, Seattle, WA, 2018.
|
|
7
|
Canani RB, Castaldo G, Bacchetta R, Martín
MG and Goulet O: Congenital diarrhoeal disorders: Advances in this
evolving web of inherited enteropathies. Nat Rev Gastroenterol
Hepatol. 12:293–302. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Goulet O, Pigneur B and Charbit-Henrion F:
Congenital enteropathies involving defects in enterocyte structure
or differentiation. Best Pract Res Clin Gastroenterol.
56-57(101784)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hao MM, Foong JP, Bornstein JC, Li ZL,
Vanden Berghe P and Boesmans W: Enteric nervous system assembly:
Functional integration within the developing gut. Dev Biol.
417:168–181. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sun Y, Leng C and van Ijzendoorn SCD:
Fetal bowel abnormalities suspected by ultrasonography in
microvillus inclusion disease: Prevalence and clinical
significance. J Clin Med. 11(4331)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Slotnick RN and Abuhamad AZ: Prognostic
implications of fetal echogenic bowel. Lancet. 347:85–87.
1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Harrison KL, Martinez D and Mason G: The
subjective assessment of echogenic fetal bowel. Ultrasound Obstet
Gynecol. 16:524–529. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lees CC, Stampalija T, Baschat A, da Silva
Costa F, Ferrazzi E, Figueras F, Hecher K, Kingdom J, Poon LC,
Salomon LJ and Unterscheider J: ISUOG Practice guidelines:
Diagnosis and management of small-for-gestational-age fetus and
fetal growth restriction. Ultrasound Obstet Gynecol. 56:298–312.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mone F, Mellis R, Gabriel H, Baptiste C,
Giordano J, Wapner R and Chitty LS: Should we offer prenatal exome
sequencing for intrauterine growth restriction or short long bones?
A systematic review and meta-analysis. Am J Obstet Gynecol.
228:409–417.e4. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Pauta M, Martinez-Portilla RJ, Meler E,
Otaño J and Borrell A: Diagnostic yield of exome sequencing in
isolated fetal growth restriction: Systematic review and
meta-analysis. Prenat Diagn. 43:596–604. 2023.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Borrell A, Grande M, Meler E, Sabrià J,
Mazarico E, Muñoz A, Rodriguez-Revenga L, Badenas C and Figueras F:
Genomic microarray in fetuses with early growth restriction: A
multicenter study. Fetal Diagn Ther. 42:174–180. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Borrell A, Grande M, Pauta M,
Rodriguez-Revenga L and Figueras F: Chromosomal microarray analysis
in fetuses with growth restriction and normal karyotype: A
systematic review and meta-analysis. Fetal Diagn Ther. 44:1–9.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Levaillant JM, Bault JP, Benoit B and
Couly G: Normal and Abnormal Fetal Face Atlas. Springer,
Switzerland, 2017.
|
|
20
|
Meler E, Sisterna S and Borrell A: Genetic
syndromes associated with isolated fetal growth restriction. Prenat
Diagn. 40:432–446. 2020.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Nowakowska BA, Pankiewicz K, Nowacka U,
Niemiec M, Kozłowski S and Issat T: Genetic background of fetal
growth restriction. Int J Mol Sci. 23(36)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Paz Y, Miño MF, Pauta M, Meler E, Matas I,
Mazarico E, Camacho A, Segura M, Figueras F and Borrell A:
Postnatal genetic and neurodevelopmental assessment in infants born
at term with severely low birth weight of non-placental origin.
Ultrasound Obstet Gynecol. 62:361–368. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
D'Amico A, Buca D, Rizzo G, Khalil A,
Silvi C, Makatsariya A, Nappi L, Liberati M and D'Antonio F:
Outcome of fetal echogenic bowel: A systematic review and
meta-analysis. Prenat Diagn. 41:391–399. 2021.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Harada A, Miyashita S, Nagai R, Makino S
and Murotsuki J: Prenatal sonographic findings and prognosis of
craniosynostosis diagnosed during the fetal and neonatal periods.
Congenit Anom (Kyoto). 59:132–141. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Miller C, Losken HW, Towbin R, Bowen A,
Mooney MP, Towbin A and Faix RS: Ultrasound diagnosis of
craniosynostosis. Cleft Palate Craniofac J. 39:73–80.
2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nagy L and Demke JC: Craniofacial
anomalies. Facial Plast Surg Clin North Am. 22:523–548.
2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Beckett JS, Pfaff MJ, Diluna M and
Steinbacher DM: Dolichocephaly without sagittal craniosynostosis. J
Craniofac Surg. 24:1713–1715. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Crystal RG: Alpha 1-antitrypsin
deficiency, emphysema, and liver disease. Genetic basis and
strategies for therapy. J Clin Invest. 85:1343–1352.
1990.PubMed/NCBI View Article : Google Scholar
|