1. Introduction
In total, >1.5 billion individuals worldwide
suffer from hearing loss, the number of which is increasing
annually (1). Apart from age, noise
is the second leading cause of hearing loss, with >500 million
individuals suffering from noise-induced hearing loss (2). At present, it is considered that
oxidative stress damage to cochlear cells, which are hair cells
(HCs) and spiral neurons, is an important cause of irreversible
noise-induced hearing loss. Emerging mechanisms, such as
mitochondrial autophagy, are beginning to garner attention
(3).
At present, the pathogenesis of various clinical
diseases, such as neurodegenerative, cardiovascular, cancer and
metabolic disorders, are gaining attention due to the role of
mitochondria as vital energy centers in eukaryotic cells.
Mitochondrial autophagy is a mechanism that maintains the stability
of the internal environment of the cell by selectively degrading
damaged mitochondria. Mechanisms, such as oxidative stress,
inflammation, apoptosis, necrosis and energy imbalances, are
closely associated with the regulation of mitochondrial autophagy
(4). In the field of studying
hearing loss mechanisms, oxidative stress is an important mechanism
that causes damage to HCs and synaptic-related diseases in the
cochlea. Mitochondrial autophagy can eliminate damaged
mitochondria, prevent the production and escape of oxidative free
radicals, which has important protective significance (5). These findings suggest that regulating
mitochondrial autophagy is closely associated with the fate of
cochlear receptor cells and therefore hearing loss.
Voltage-dependent anion channel 1 (VDAC1) is a key mitochondrial
outer membrane protein that is associated with the PTEN-induced
putative kinase 1 (PINK1)/Parkin pathway (6). VDAC1 serves a role in maintaining the
stability of the mitochondrial outer membrane (7). It is associated with the release of
mitochondrial apoptosis necrosis factors [such as
apoptosis-inducing factor (AIF) and cytochrome c (cyto c)],
ions and mitochondrial DNA (mtDNA) (8), serving a dual role in determining cell
fate within signaling pathways. It can be hypothesized that this
may be a key molecule in regulating the balance between
mitochondrial autophagy and cell death. The present review
summarizes and examines the relevant literature on VDAC1 and its
role, in addition to surmising its clinical translational
significance in hearing loss research.
2. Mitophagy and oxidative stress in hearing
loss
Hearing loss is a condition in which external
stimuli causes damage to the sensory cells in the cochlea,
resulting in impaired sound transmission. Ageing, noise, infections
and ototoxic drugs can all lead to sensory cell damage. Permanent
hearing loss can result from irreversible sensory cell damage, such
as the destruction of cochlear hair cells due to noise-induced
oxidative stress (9). However,
there are cases of neurological hearing loss in clinical practice
where hearing is restored through intervention measures during the
early stages of injury (10),
indicating the existence of mechanisms regulating the death and
survival of cochlear sensory cells. Mitochondrial autophagy has
been recognized to be an important endogenous mechanism for
maintaining mitochondrial quality.
Mitochondrial autophagy is a vital intrinsic
protective mechanism that enables cells to respond to stimuli and
damage, significantly contributing to the process of hearing loss.
Oxidative stress leads to mitochondrial dysfunction, triggering
autophagy reactions to clear damaged mitochondria and excess
reactive oxygen species (ROS) (11). ROS are mainly derived from
mitochondria (12). Superoxide
dismutase (SOD)2 in the mitochondrial matrix and SOD1 in the
intermembrane space catalyze the conversion of
O2- to H2O2, which
subsequently transforms into superoxide (13). The destructive effects of ROS have
been extensively studied. Morioka et al (9) previously found that excessive ROS can
damage inner ear hair cells, resulting in hearing loss. Oxidative
stress is a key pathogenic factor in the noise-induced hearing loss
(NIHL) model. Liang et al (14) reported that ROS accumulation in the
inner ear is significantly associated with cell death following
noise exposure. However, ROS can also induce autophagy (15). A previous study revealed that the
activation of autophagy in outer hair cells (OHCs) can exert a
protective effect against NIHL by reducing oxidative stress
(16). During noise-induced
temporary threshold shift, oxidative stress can elevate
microtubule-associated protein 1 light chain 3B (LC3B) levels in
OHCs, while sirolimus treatment can mitigate 4-hydroxynonenal and
3-nitrotyrosine (3-NT) levels to reduce OHC damage. Similarly, the
use of 3-methyladenine or LC3B small interfering RNA leads to an
increase in 3-NT levels and cell death in OHCs (17).
Therefore, cell fate following damage caused by ROS
to the cochlear sensory cells depends on the balance achieved by
mitochondrial autophagy and apoptosis signalling pathways. The
apoptosis-related pathways guided by mitochondria, such as caspase
pathway and AIF pathway, involve the release of apoptosis factors
from mitochondria, such as the TNF receptor (18). In addition, mitochondrial autophagy,
as a cellular protective pathway, can also promote the clearance of
damaged mitochondria and maintain mitochondrial function through
PINK/Parkin-mediated ubiquitination (19). However, during the temporary
threshold shift, the increased expression of mitophagy-related
protein LC3B, activated by oxidative stress, indicates that
oxidative stress also promotes mitochondrial autophagy, as
previously reported (19). Overall,
mitochondrial autophagy and apoptosis may function as balanced
regulators of cell fate, however there is no literature that
provides a detailed explanation on how the balance between
autophagy and apoptosis is regulated. A study of mitochondrial
autophagy previously revealed that VDAC1 is involved in cell damage
and can mediate a dual regulatory effect. It primarily serves a
role in regulating the mitochondrial permeability transition pore
(20), facilitating the movement of
apoptotic factors (such as AIF) from the mitochondria to the
cytoplasm, thereby contributing to cell apoptosis (21). It also functions as a regulator of
mitochondrial autophagy by recruiting the autophagy receptor
protein sequestosome 1 (SQSTM1/p62), thereby mediating
mitochondrial autophagy to maintain mitochondrial quality control
and cell survival balance (22).
Huang et al (23)
demonstrated the interaction between VDAC1 and myeloid cell
leukemia 1 as being crucial for mitochondrial function and ROS
regulation, offering insights into hearing loss pathogenesis. In
another study, Kurabi et al (5) highlighted the association between cell
apoptosis and inner ear damage following noise exposure, noting the
involvement of VDAC1 underlying this apoptotic process. The
interplay between autophagy and oxidative stress is likely
important in hearing loss, with the bidirectional functionality of
VDAC1 being potentially pivotal.
3. Molecular characteristics and dual
regulatory mechanism of VDAC1
Structural features and basic
functions
VDAC1 is a key protein on the mitochondrial outer
membrane that serves to regulate substance and ion exchange between
mitochondria and the cytoplasm upstream of cellular energy
metabolism. It participates in various functions, such as energy
generation, calcium ion transport and the regulation of cell
apoptosis (24). Research has
indicated that VDAC1 expression can vary in neurodegenerative
diseases (such as Alzheimer's disease), in addition to interacting
with disease-related molecules and contributing to disease
progression (25). VDAC was
initially purified from paramecium mitochondria in
1976(25). At present, three
subtypes of VDAC family proteins have been identified in mammals,
namely VDAC1, VDAC2 and VDAC3. VDAC1 is extensively expressed and
exhibits notable bidirectional functionality (24,26).
VDAC2 has been reported to interact with proteins containing Bcl-2
homology 3 (BH3) death domains, including truncated BH3-interacting
domain death agonist, Bcl-2-like protein 11 and and
Bcl-2-associated agonist of cell death, to facilitate Bcl-2
homologous antagonist/killer oligomerization, create apoptotic
channels and trigger cell apoptosis (27). VDAC3, which is particularly rich in
cysteine residues, is important for mitochondrial protection
(28). VDAC subtypes have a
tissue-specific expression pattern, where VDAC1 is highly expressed
in general, especially in the cochlea. VDAC1 is the most highly
expressed among the three subtypes in most organs, such as the
heart, liver, brain tissue, including cochlear tissue and other
mitochondrial-rich tissues. VDAC1 is considered the gatekeeper of
mitochondrial and cytoplasmic material transport, and is therefore
widely expressed in mitochondrial-rich tissues (29). In the cochlea (30), hair cells and spiral neurons are the
concentrated areas of mitochondrial distribution, therefore, VDAC1
exhibits expression distribution in this region. In addition, in
the latest research on the mechanism of hearing loss (31), it was found that VDAC1 is widely
expressed in cochlear cells and increases after noise exposure.
VDAC1 consists of 19 transmembrane β chains forming
a β pore, which contains a 25-residue fragment in the N-terminal
domain. N-terminal domain migration serves a role in channel gating
and VDAC1 dimer formation. An α-helix is located at the midpoint of
the barrel hole and is horizontal, where it regulates the transport
of metabolites by reducing the pore size (32). VDAC1 facilitates the transfer of a
number of metabolites, such as pyruvate, succinate, malonate,
nucleotides and NADH, into the mitochondria, enabling subsequent
metabolism (33). In addition,
VDAC1 is involved in cholesterol transport, lipid metabolism,
Ca2+ signaling between mitochondria and the endoplasmic
reticulum and redox status regulation (34). VDAC1 is also essential for
mitochondria-mediated cell apoptosis. Drug-induced cell apoptosis
can promote overexpression and oligomerization of VDAC1, leading to
the formation of large pores on the surface of mitochondria, and
promoting the transfer of AIF, cyto c and apoptotic
protease-activating factor 1 (Apaf-1) to the cytoplasm, thereby
activating apoptotic pathways (35-37).
For example, AIF can transfer out of mitochondria and enter the
nucleus to promote DNA condensation and cleavage, and promote cell
apoptosis. Similarly, cyto c and Apaf-1 are also involved in
related apoptotic mechanisms. Therefore, VDAC1 is considered a
‘mitochondrial gatekeeper’.
Ubiquitination modification and
mitophagy regulation
VDAC1 regulates cell fate determination through the
dynamic balance of the ubiquitination/oligomerization switch.
Through the classical pathway, VDAC1 serves as a substrate for the
E3 ligase Parkin (38). During
autophagy induction, VDAC1 can be ubiquitinated to form adapter
proteins with ubiquitin-binding motifs and LC3B-binding motifs,
recruiting p62/SQSTM1 or LC3B to promote autophagosome formation
and induce lysosomal binding to these labelled mitochondria for
selective degradation. In the traditional PINK1/Parkin
mitochondrial autophagy pathway, PINK1 phosphorylates Parkin on S65
within the ubiquitin-like domain, thereby activating its E3
ubiquitin ligase activity. Notable Parkin substrates include the
mitochondrial outer membrane-associated proteins mitofusin (MFN)1,
MFN2, outer membrane transposase 20, mitochondrial Rho GTPase 1 and
VDAC1, which clear damaged mitochondria and maintain their
stability through the ubiquitination autophagy mechanism (39). In addition, the ubiquitination sites
of VDAC1 include β-chain K53, K274, K12, K20, K53, K109 and K110.
In a previous study on liver fibrosis, ubiquitination at the K53
site of VDAC1 was found to negatively regulate VDAC1
oligomerization and mtDNA release (40). Apart from Parkin, other E3 ubiquitin
ligases, such as neural precursor cell expression and developmental
downregulation 4, can connect with the ubiquitylated protein
through the K48 site to negatively regulate acetaminophen-induced
mitochondrial damage in liver cells (41). In Parkinson's disease, tripartite
motif-containing protein 31, a triple motif protein, can promote
the degradation of VDAC1 by catalyzing ubiquitination at the K48
site of VDAC1, a process which helps stabilize dopaminergic neurons
(42). Therefore, investigating the
ubiquitination sites and specific signaling pathways of VDAC1 in
cochlear receptor cells offers a novel perspective for studying
hearing loss mechanisms.
Oligomerization modification and
activation of cell apoptosis
There are multiple post-translational modification
modes of VDAC1, including phosphorylation, acetylation, tyrosine
nitration, ubiquitination and oligomerization, but their
consequences remain poorly elucidated. VDAC1 is known for its
oligomerization, because it is associated with cell death. VDAC1
can form pore-like channels in the outer membrane of mitochondria,
regulating the entry and exit of Ca2+ to maintain the
balance of the outer membrane. However, under stimulation, VDAC1
will self-aggregate to form dimeric, trimeric or even oligomeric
forms, expanding the pore size and disrupting the balance on the
outer membrane of mitochondria. During cell apoptosis, it interacts
with the apoptotic protein Bax to form an oligomeric structure that
increases the central pore size of VDAC1, allowing the release of
cyto c, Apaf-1, deoxyadenosine triphopshate (dATP) and AIF to
promote apoptosis (35). VDAC1
oligomerization pores also facilitate mtDNA release from
mitochondria into the cytoplasm, disrupting mitochondrial
homeostasis and aggravating lupus erythematosus (43). In addition, VDAC1 is associated with
the uptake and release of mitochondrial Ca2+. It can
interact with the endoplasmic reticulum and become an important
part of the mitochondrial Ca2+ connection. Excessive
Ca2+ transport can also stimulate the regulatory effect
of VDAC1(44). A previous study
reported that upregulation of the mitochondrial calcium uniporter
protein in a cadmium toxicity model of liver cells can trigger the
oligomerization and ubiquitination of VDAC1 (45,46),
where the balance between oligomerization and ubiquitination can
determine the fate of cells.
VDAC1 plays a significant role in
ubiquitination
Mitochondrial autophagy involves a critical step
known as ubiquitination, which functions analogously to tagging
substrates for subsequent lysosomal degradation. While
ubiquitination marks cellular components for phagocytosis by
lysosomes, the detailed mechanisms of lysosomal engulfment and
degradation fall beyond the scope of this discussion (42). Previous research has elucidated the
role of VDAC1 in mitochondrial ubiquitination, such as its
potential as a substrate for E3 ubiquitin ligase (47). It can also regulate classic
mitochondrial autophagy pathways such as the PINK1/Parkin pathway.
However, the role of VADC1 in autophagy remains only partially
understood, including its inhibition of the PINK1/Parkin pathway to
mitigate noise-induced hearing loss (31). In addition, increased expression of
VDAC1 may serve as an additional anchor site for polyubiquitination
recruitment, triggering mitochondrial autophagy (48). Thus, VDAC1 has been revealed to be
associated with autophagy, but further research is still
required.
4. Possible mechanism of VDAC1 in hearing
loss
At present, research on the mechanism of hearing
loss injury caused by VDAC1 remains insufficient and is limited
mainly to its regulatory effect on Ca2+. El-Emam et
al (49) found that the
inositol trisphosphate receptor/glucose-regulated protein 75/VDAC1
complex in cochlear sensory cell mitochondria can facilitate the
transfer of Ca2+ from the endoplasmic reticulum into the
mitochondria. In another study, Zhao et al (50) reported that endoplasmic reticulum
Ca2+ can transfer to the mitochondria through the VDAC1
pathway, triggering autophagy in HEI-OC1 cells in response to
cisplatin (50). Li et al
(51) proposed that the decline in
mitochondrial function associated with noise-induced hearing loss
is associated with oxidative stress and nucleotide-binding
oligomerization domain-, leucine-rich repeat-, and pyrin
domain-containing protein 3 inflammasome activation, with VDAC1
involved in mitochondrial regulation (51). Wang et al (52) previously found that VDAC1 expression
is significantly increased in a noise-induced hearing loss model,
which was associated with mitochondrial dysfunction (52). Le et al (53) emphasized the impact of noise-induced
hearing loss on cochlear cells, highlighting the importance of
mitochondrial membrane stability (53). A previous study demonstrated that
inhibition of VDAC1 can enhance mitochondrial autophagy in cochlear
hair cells within cochlear explants and HEI-OC1 cells, thereby
serving a key role in mitigating oxidative stress and inflammation
(50). Downregulation of VDAC1 has
been found to reduce hair cell damage and apoptosis by activating
the PINK1/Parkin pathway, thereby contributing to the preservation
of hearing function (31). However,
the multiphase protein modification and regulatory mechanisms of
VDAC1 in sensory cell damage in individuals with hearing loss has
not been fully elucidated. According to a previous study, after 7
days of noise exposure, the expression of AIF decreased in cochlear
sensory cells suggesting a potential protective response to
mitochondrial dysfunction (54). Ma
et al (47) reported that
the decrease in AIF expression during hypoxia is due to the
ubiquitination and clearance of ubiquitin A-52 residue ribosomal
protein fusion product 1 (UBA52) by ubiquitination. Tiwari et
al (55) reported that UBA52
can also ubiquitinate VDAC1 through the E3 ligase carboxyl terminus
of Hsc70-interacting protein, thereby activating mitochondrial
autophagy. These findings establish a link between VDAC1
ubiquitination and AIF ubiquitination clearance. In addition, Chen
et al (38) reported that
VDAC1 expression was reduced to activate autophagy and alleviate
cell apoptosis in leafhoppers infected with arboviruses, thereby
protecting the cells. These findings indicate that VDAC1
ubiquitination can reduce cell apoptosis and regulate autophagy
(38). In addition, the
aforementioned findings offer insights into the study of
noise-induced hearing loss and its prevention (Fig. 1). Further elucidation of the role of
VDAC1 in the regulation of cell survival and death is key to
identifying novel targets for cochlear sensory cell protection.
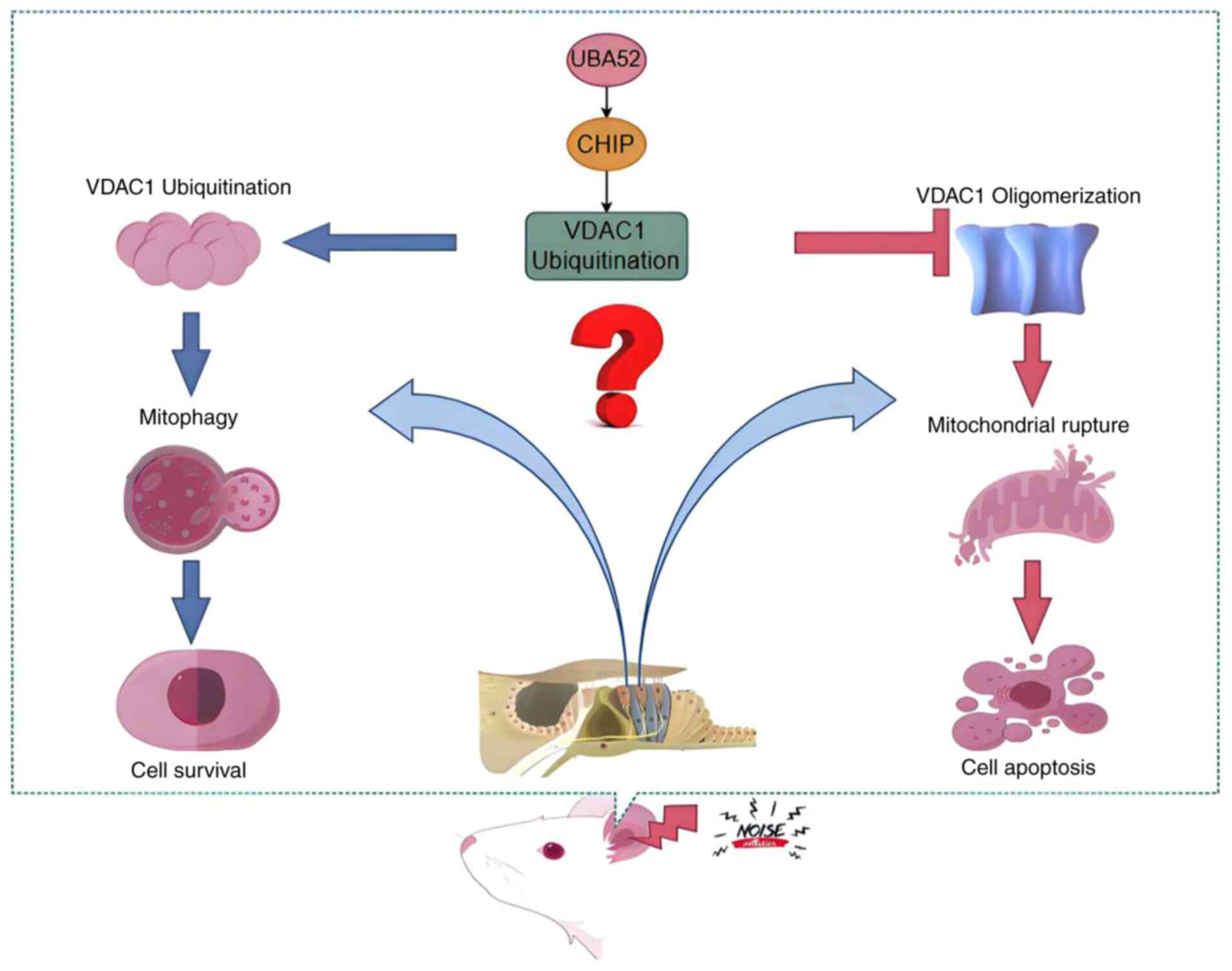 | Figure 1Hypothetical diagram illustrating the
effect of VDAC1 on the fate of cochlear cells under noise
stimulation. The regulation of VDAC1 ubiquitination by the E3
ubiquitin ligase CHIP may lead to different fates of cochlear
sensory cells. First, increased VDAC1 ubiquitination leads to
mitochondrial autophagy, and mitochondrial quality control mediates
the clearance of damage factors and cell survival. Second,
decreased VDAC1 ubiquitination mediates VDAC1 oligomerization in
the outer mitochondrial membrane, leading to abnormal opening of
the mitochondrial permeability transition pore, the release of
apoptotic factors, mitochondrial rupture, and cell apoptosis. The
figure was created using Figdraw 2.0 Online Drawing Platform;
https://www.figdraw.com/#/. VDAC1,
voltage-dependent anion channel 1; CHIP, carboxy terminus of
HSP-70-interacting protein; UBA52, ubiquitin A-52 residue ribosomal
protein fusion product 1. |
Prospects and clinical translation of
VDAC1 in hearing loss
Given the potential role of VDAC1 in cochlear
sensory cell stress, modulating VDAC1 function can enhance cochlear
cell environments, mitigate oxidative stress and bolster
resistance, thereby reducing cochlear cell damage. Livingston et
al (56) previously showed that
ischemic pretreatment of renal tubular cells activated autophagy,
which prevented mitochondrial depolarization, enhanced ATP
production and regulated cell apoptosis and autophagy, thereby
protecting these cells from ischemic damage and offering novel
insights for hearing loss treatment. It is surmised that reducing
the expression of VDAC1, regulating VDAC1 ubiquitination, enhancing
mitochondrial autophagy, reducing cell damage and protecting
hearing are effective strategies in protecting from hearing loss.
The VDAC1 oligomerization inhibitor, VBIT-4, has been previously
applied to reduce mtDNA release by inhibiting VDAC1
oligomerization, which had protective significance in lupus-like
diseases (57). VBIT-4 has also
been reported to alleviate the neuropathological features of
Alzheimer's disease mouse models by targeting VDAC1(58). In addition, VDAC1 inhibition has
been achieved using CRISPR/Cas9 gene editing technology to alter
its active site (59). However,
this approach is currently limited to scientific research, where
owing to the biological homology of VDAC1, knockout has not
achieved protective results. In addition, the clinical use of the
antidepressant sertraline (Sert) has been reported to act as an
autophagy inducer, targeting and antagonizing VDAC1 expression and
enhancing autophagy (60).
Exploring the application of Sert as a protective measure against
noise-induced hearing loss presents a potential intervention
strategy. Consequently, targeting VDAC1 may offer a novel approach
for future hearing loss treatments. It is necessary to further
explore the mechanism of VDAC1 protein modification in different
types of sensory cells involved in hearing loss, such as hair cells
and spiral neurons, to integrate the protein modification
strategies of VDAC1 and provide theoretical evidence for research
on hearing protection.
In summary, there is a possible association between
VDAC1 and hearing loss through mitochondrial autophagy, but there
are unsolved research gaps. Further exploration and research are
needed to determine which sensory cells are regulated by VDAC1
under conditions of hearing loss injury and whether this regulation
can induce mitochondrial autophagy or cell death. Furthermore,
little is known regarding the specific regulatory mechanism of
targets in the ubiquitination process of VDAC1. The advancement in
VDAC1 inhibitors for Alzheimer's and type 2 diabetes, along with
the significance of ubiquitination regulation in drug development,
offers a novel approach for future hearing loss treatment and
prevention.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
ZJD contributed to the conception, formal analysis,
and supervision of the study. Additionally, ZJD played a key role
in writing the original draft and participated in the review and
editing process. YW contributed to the writing, reviewing and
editing of the manuscript, as well as providing consultation. Both
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen F, Xue H, Wang M, Cai Z and Zhu S:
Hearing care: Safe listening method and system for personal
listening devices. Int J Environ Res Public Health.
20(2161)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Maniaci A, La Via L, Lechien JR,
Sangiorgio G, Iannella G, Magliulo G, Pace A, Mat Q, Lavalle S and
Lentini M: Hearing loss and oxidative stress: A comprehensive
review. Antioxidants (Basel). 13(842)2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang Y, Fang Q, Wang H, Qi J, Sun S, Liao
M, Wu Y, Hu Y, Jiang P, Cheng C, et al: Increased mitophagy
protects cochlear hair cells from aminoglycoside-induced damage.
Autophagy. 19:75–91. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Baechler BL, Bloemberg D and Quadrilatero
J: Mitophagy regulates mitochondrial network signaling, oxidative
stress, and apoptosis during myoblast differentiation. Autophagy.
15:1606–1619. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kurabi A, Keithley EM, Housley GD, Ryan AF
and Wong AC: Cellular mechanisms of noise-induced hearing loss.
Hear Res. 349:129–137. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Geula S, Ben-Hail D and Shoshan-Barmatz V:
Structure-based analysis of VDAC1: N-terminus location,
translocation, channel gating and association with anti-apoptotic
proteins. Biochem J. 444:475–485. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vijayan M, Alvir RV, Alvir RV, Bunquin LE,
Pradeepkiran JA and Reddy PH: A partial reduction of VDAC1 enhances
mitophagy, autophagy, synaptic activities in a transgenic Tau mouse
model. Aging Cell. 21(13663)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Shoshan-Barmatz V, Shteinfer-Kuzmine A and
Verma A: VDAC1 at the intersection of cell metabolism, apoptosis,
and diseases. Biomolecules. 10(1485)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Morioka S, Sakaguchi H, Yamaguchi T,
Ninoyu Y, Mohri H, Nakamura T, Hisa Y, Ogita K, Saito N and Ueyama
T: Hearing vulnerability after noise exposure in a mouse model of
reactive oxygen species overproduction. J Neurochem. 146:459–473.
2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Young Y: Contemporary review of the causes
and differential diagnosis of sudden sensorineural hearing loss.
Int J Audiol. 59:243–253. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fetoni AR, De Bartolo P, Eramo SLM, Rolesi
R, Paciello F, Bergamini C, Fato R, Paludetti G, Petrosini L and
Troiani D: Noise-induced hearing loss (NIHL) as a target of
oxidative stress-mediated damage: Cochlear and cortical responses
after an increase in antioxidant defense. J Neurosci. 33:4011–4023.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tretter V, Hochreiter B, Zach ML, Krenn K
and Klein KU: Understanding cellular redox homeostasis: A challenge
for precision medicine. Int J Mol Sci. 23(106)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Adam-Vizi V: Production of reactive oxygen
species in brain mitochondria: Contribution by electron transport
chain and non-electron transport chain sources. Antioxid Redox
Signal. 7:1140–1149. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liang S, Dong S, Liu W, Wang M, Tian S, Ai
Y and Wang H: Accumulated ROS activates HIF-1α-induced glycolysis
and exerts a protective effect on sensory hair cells against
noise-induced damage. Front Mol Biosci. 8(806650)2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Navarro-Yepes J, Burns M, Anandhan A,
Khalimonchuk O, Del Razo LM, Quintanilla-Vega B, Pappa A,
Panayiotidis MI and Franco R: Oxidative stress, redox signaling,
and autophagy: Cell death versus survival. Antioxid Redox Signal.
21:66–85. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li Y, Li S, Wu L, Wu T, Li M, Du D, Chen
Y, Wang C, Li X, Zhang S, et al: Sestrin 2 deficiency exacerbates
noise-induced cochlear injury through inhibiting
ULK1/Parkin-mediated mitophagy. Antioxid Redox Signal. 38:115–136.
2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yuan H, Wang X, Hill K, Chen J, Lemasters
J, Yang SM and Sha SH: Autophagy attenuates noise-induced hearing
loss by reducing oxidative stress. Antioxid Redox Signal.
22:1308–1324. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chu Q, Gu X, Zheng Q, Wang J and Zhu H:
Mitochondrial mechanisms of apoptosis and necroptosis in liver
diseases. Anal Cell Pathol (Amst). 2021(8900122)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jia K and Du H: Mitochondrial permeability
transition: A pore intertwines brain aging and Alzheimer's disease.
Cells. 10(649)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu C, Wei Q, Li X, Han D, Liu J, Huang F
and Zhang C: Proteomic analyses of mitochondrial damage in
postmortem beef muscles. J Sci Food Agric. 102:4182–4191.
2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Huang H, Shah K, Bradbury NA, Li C and
White C: Mcl-1 promotes lung cancer cell migration by directly
interacting with VDAC to increase mitochondrial Ca2+ uptake and
reactive oxygen species generation. Cell Death Dis.
5(e1482)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Di Rosa MC, Guarino F, Conti Nibali S,
Magri A and De Pinto V: Voltage-dependent anion selective channel
isoforms in yeast: Expression, structure, and functions. Front
Physiol. 12(675708)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Schein SJ, Colombini M and Finkelstein A:
Reconstitution in planar lipid bilayers of a voltage-dependent
anion-selective channel obtained from paramecium mitochondria. J
Membr Biol. 30:99–120. 1976.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zinghirino F, Pappalardo XG, Messina A,
Guarino F and De Pinto V: Is the secret of VDAC Isoforms in their
gene regulation? Characterization of human VDAC genes expression
profile, promoter activity, and transcriptional regulators. Int J
Mol Sci. 21(7388)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lalier L, Cartron P, Juin P, Nedelkina S,
Manon S, Bechinger B and Vallette FM: Bax activation and
mitochondrial insertion during apoptosis. Apoptosis. 12:887–896.
2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Reina S, Nibali SC, Tomasello MF, Magri A,
Messina A and De Pinto V: Voltage dependent anion channel 3 (VDAC3)
protects mitochondria from oxidative stress. Redox Biol.
51(102264)2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Messina A, Reina S, Guarino F and De Pinto
V: VDAC isoforms in mammals. Biochim Biophys Acta. 1818:1466–1476.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lysakowski A, Govindaraju AC and Raphael
RM: Structural and functional diversity of mitochondria in
vestibular/cochlear hair cells and vestibular calyx afferents. Hear
Res. 426(108612)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jin Y, Dong W, Jiang Y, Dong L, Li Z and
Yu D: Vdac1 inhibition protects against noise-induced hearing loss
via the pink1/Parkin pathway. CNS Neurosci Ther.
31(e70410)2025.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hiller S, Abramson J, Mannella C, Wagner G
and Zeth K: The 3D structures of VDAC represent a native
conformation. Trends Biochem Sci. 35:514–521. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shoshan-Barmatz V, Maldonado EN and Krelin
Y: VDAC1 at the crossroads of cell metabolism, apoptosis and cell
stress. Cell Stress. 1:11–36. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Keinan N, Tyomkin D and Shoshan-Barmatz V:
Oligomerization of the mitochondrial protein voltage-dependent
anion channel is coupled to the induction of apoptosis. Mol Cell
Biol. 30:5698–5709. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zalk R, Israelson A, Garty ES,
Azoulay-Zohar H and Shoshan-Barmatz V: Oligomeric states of the
voltage-dependent anion channel and cytochrome c release from
mitochondria. Biochem J. 386:73–83. 2005.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Aram L, Geula S, Arbel N and
Shoshan-Barmatz V: VDAC1 cysteine residues: topology and function
in channel activity and apoptosis. Biochem J. 427:445–454.
2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen Q, Jia D, Ren J, Cheng Y, Wu H, Guo S
and Wei T: VDAC1 balances mitophagy and apoptosis in leafhopper
upon arbovirus infection. Autophagy. 19:1678–1692. 2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li J, Yang D, Li Z, Zhao M, Wang D, Sun Z,
Wen P, Dai Y, Gou F, Ji Y, et al: PINK1/Parkin-mediated mitophagy
in neurodegenerative diseases. Ageing Res Rev.
84(101817)2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wu NN, Wang L, Wang L, Xu X, Lopaschuk GD,
Zhang Y and Ren J: Site-specific ubiquitination of VDAC1 restricts
its oligomerization and mitochondrial DNA release in liver
fibrosis. Exp Mol Med. 55:269–280. 2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu Y, Lei L, Wang X, Chen L, Li W, Li J,
Zhao C, Du X, Song Y, Gao W, et al: The E3 ubiquitin ligase NEDD4-1
protects against acetaminophen-induced liver injury by targeting
VDAC1 for degradation. Acta Pharm Sin B. 13:1616–1630.
2023.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhao Z, Song X, Wang Y, Yu L, Huang G, Li
Y, Zong R, Liu T, Ji Q, Zheng Y, et al: E3 ubiquitin ligase TRIM31
alleviates dopaminergic neurodegeneration by promoting proteasomal
degradation of VDAC1 in Parkinson's disease model. Cell Death
Differ. 31:1410–1421. 2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xian H, Watari K, Sanchez-Lopez E,
Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS
and Karin M: Oxidized DNA fragments exit mitochondria via mPTP- and
VDAC-dependent channels to activate NLRP3 inflammasome and
interferon signaling. Immunity. 55:1370–1385.e8. 2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Huang H, Hu X, Eno CO, Zhao G, Li C and
White C: An interaction between Bcl-xL and the voltage-dependent
anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol
Chem. 288:19870–19881. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Liu C, Li HJ, Duan WX, Duan Y, Yu Q, Zhang
T, Sun YP, Li YY, Liu YS and Xu SC: MCU upregulation overactivates
mitophagy by promoting VDAC1 dimerization and ubiquitination in the
hepatotoxicity of cadmium. Adv Sci (Weinh).
10(e2203869)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ham SJ, Lee D, Yoo H, Jun K, Shin H and
Chung J: Decision between mitophagy and apoptosis by Parkin via
VDAC1 ubiquitination. Proc Natl Acad Sci USA. 117:4281–4291.
2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ma C, Wang X, He S, Zhang L, Bai J, Qu L,
Qi J, Zheng X, Zhu X, Mei J, et al: Ubiquitinated AIF is a major
mediator of hypoxia-induced mitochondrial dysfunction and pulmonary
artery smooth muscle cell proliferation. Cell Biosci.
12(9)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Liu Y, Zhang H, Liu Y, Zhang S, Su P, Wang
L, Li Y, Liang Y, Wang X, Zhao W, et al: Hypoxia-induced GPCPD1
depalmitoylation triggers mitophagy via regulating PRKN-mediated
ubiquitination of VDAC1. Autophagy. 19:2443–2463. 2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
El-Emam MA, Sheta E, El-Abhar HS, Abdallah
DM, El Kerdawy AM, Eldehna WM and Gowayed MA: Morin suppresses
mTORc1/IRE-1α/JNK and IP3R-VDAC-1 pathways: Crucial mechanisms in
apoptosis and mitophagy inhibition in experimental Huntington's
disease, supported by in silico molecular docking simulations. Life
Sci. 338(122362)2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhao H, Xu Y, Song X, Zhang Q, Wang Y, Yin
H, Bai X and Li J: Cisplatin induces damage of auditory cells:
Possible relation with dynamic variation in calcium homeostasis and
responding channels. Eur J Pharmacol. 914(174662)2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Li P, Li S, Wang L, Li H, Wang Y, Liu H,
Wang X, Zhu X, Liu Z, Ye F and Zhang Y: Mitochondrial dysfunction
in hearing loss: Oxidative stress, autophagy and NLRP3
inflammasome. Front Cell Dev Biol. 11(1119773)2023.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wang X, Zhu Y, Long H, Pan S, Xiong H,
Fang Q, Hill K, Lai R, Yuan H and Sha S: Mitochondrial calcium
transporters mediate sensitivity to noise-induced losses of hair
cells and cochlear synapses. Front Mol Neurosci.
11(469)2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Le TN, Straatman LV, Lea J and Westerberg
B: Current insights in noise-induced hearing loss: A literature
review of the underlying mechanism, pathophysiology, asymmetry, and
management options. J Otolaryngol Head Neck Surg.
46(41)2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zhong-Jia D, Ren-Feng W, Wen-Juan M, Yin W
and Ding-Jun Z: Calpain2-mediated downregulation of
apoptosis-inducing factors impairs mitochondrial function in
noise-induced spiral ganglion neuron degeneration. IBRO Neurosci
Rep. 19:372–380. 2025.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Tiwari S, Singh A, Gupta P, K A and Singh
S: UBA52 attunes VDAC1-mediated mitochondrial dysfunction and
dopaminergic neuronal death. ACS Chem Neurosci. 14:839–850.
2023.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Livingston MJ, Wang J, Zhou J, Wu G,
Ganley IG, Hill JA, Yin X and Dong Z: Clearance of damaged
mitochondria via mitophagy is important to the protective effect of
ischemic preconditioning in kidneys. Autophagy. 15:2142–2162.
2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Kim J, Gupta R, Blanco LP, Yang S,
Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, et
al: VDAC oligomers form mitochondrial pores to release mtDNA
fragments and promote lupus-like disease. Science. 366:1531–1536.
2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Verma A, Shteinfer-Kuzmine A, Kamenetsky
N, Pittala S, Paul A, Nahon Crystal E, Ouro A, Chalifa-Caspi V,
Pandey SK, Monsonego A, et al: Targeting the overexpressed
mitochondrial protein VDAC1 in a mouse model of Alzheimer's disease
protects against mitochondrial dysfunction and mitigates brain
pathology. Transl Neurodegener. 11(58)2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Yang M, Sun J, Stowe DF, Tajkhorshid E,
Kwok W and Camara AKS: Knockout of VDAC1 in H9c2 cells promotes
oxidative stress-induced cell apoptosis through decreased
mitochondrial hexokinase II binding and enhanced glycolytic stress.
Cell Physiol Biochem. 54:853–874. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hwang H, Shim JS, Kim D and Kwon HJ:
Antidepressant drug sertraline modulates AMPK-MTOR
signaling-mediated autophagy via targeting mitochondrial VDAC1
protein. Autophagy. 17:2783–2799. 2021.PubMed/NCBI View Article : Google Scholar
|














