Introduction
Waardenburg syndrome (WS) consists of a group of
four genetic disorders (WS1, OMIM#193500; WS2, OMIM#193510; WS3,
OMIM#148820 and WS4, OMIM#277580; www.omim.org),
which were initially described in 1951 by the Dutch ophthalmologist
and geneticist Petrus Johannes Waardenburg, after whom WS was
named. The prevalence varies throughout the world. A general
estimate of the syndrome is ~1 case/42,000 individuals worldwide.
The types of the syndrome are characterized by variable features in
affected individuals, including hearing loss (HL), ophthalmological
abnormalities and pigmentation disorders, as well as facial
dysmorphism (1).
WS is caused by mutations in various genes that
affect the function of nerve cells in embryonic development. The
majority of the types are caused by autosomal dominant mutations.
Autosomal recessive mutations are few and rare. In the majority of
the cases, the affected individual inherits WS from a parent with
one of the dominant forms of the condition. A low percentage of
cases result from spontaneous new mutations (de novo) in the
gene, where there is no family history of the condition. WS is
usually caused by mutations in the genes paired box 3
(PAX3), Endothelin 3 (EDN3), Endothelin Receptor Type
B (EDNRB), Melanocyte Inducing Transcription Factor
(MITF), Snail Family Transcriptional Repressor 2
(SNAI2), SRY-Box Transcription Factor 10 (SOX10)
(2). Mutations in the pax3
gene are associated with WS1 and WS3, which are caused in the
majority of the cases by heterozygous pathogenic mutations, while
WS3 has been reported as a result of homozygous pathogenic
mutations with more severe manifestations (3,4). WS2
usually occurs from pathogenic heterozygous mutations in the
MITF, SOX10 and EDNRB genes (2,5), and
in the KITLG gene in heterozygous and homozygous forms
(6,7). Overall, ~50% of WS4 cases are caused
by heterozygous pathogenic mutations in sox10, whereas
20-30% of the cases are caused by pathogenic mutations in
EDNRB and EDN3, with dominant/recessive forms of
inheritance (8-10).
In humans the chromosomal locus of the PAX3
gene is Chr2q36.1, from 222.199.888 to 222.298.996 bp in a 100-kb
region (Ensembl assembly release GRCh38.p10). The PAX3 gene
includes 10 exons; more specifically exons 2, 3 and 4 encode the
paired box (PB), 5 and 6 encode the conserved octopeptide and
homeodomain (HD), and 7 and 8 encode the activation region
(11). At the mRNA level, at least
10 different pax3 isoforms have been detected as a consequence of
alternative splicing and processing. The longest isoform appears to
be pax3e, consisting of 10 exons, which encodes a 505-amino acid
protein (12). The expression of
the protein occurs during the formation of the central nervous
system, skeletal muscle, neural crest and derivatives. The pax3
protein operates as a regulator of target gene expression affecting
differentiation, survival, migration, motility and proliferation in
these lineages (13). The pax3
protein is a transcription factor, which consists of the two
following functional domains: An N-terminal DNA-binding region that
includes the PB, octapeptide and HD, and a C-terminal transcription
activation region that includes a proline, serine and
threonine-rich site.
The diagnosis of WS can often be difficult due to
the wide range of symptoms and genetic variants. The condition can
be diagnosed before/at birth, in early childhood or in certain
cases at an older age with a comprehensive clinical evaluation,
identification of clinical features, a detailed patient and family
history, and several specialized genetic tests. A detailed medical
evaluation and genetic testing are necessary to determine the
presence of the condition and select appropriate supportive and
symptomatic treatments (14).
The present study describes the case of a
33-year-old male with clinical features compatible with WS
undergoing genomic testing by next generation sequencing (NGS). NGS
testing identified a novel c.209G>A (p.Cys70Tyr) mutation in the
PAX3 gene, which was likely responsible for the observed
pathogenesis of the syndrome. The aim of the present study is to
investigate the association between the syndrome and mutations in
the PAX3 gene. The existing international literature is
analyzed and the documented clinical cases are presented to provide
an improved understanding of the role of the PAX3 gene in
the pathogenesis of the syndrome. The present study is important as
it identifies a novel mutation that may contribute to more
extensive genetic counseling and improved diagnosis and management
of WS.
Case report
The present study is a case report of a 33-year-old
male, who after a detailed clinical evaluation, identification of
characteristic physical findings, and analysis of a detailed
patient and family history, was found to have clinical features
compatible with Waardenburg syndrome (WS). In particular, the
patient had undergone regular hearing tests, which revealed
bilateral neurosensory HL bordering on practical deafness and a
hearing impairment rate of >67%. This clinical presentation was
established during infancy and oral speech disorders had since
occurred. In addition, following a medical examination, it was
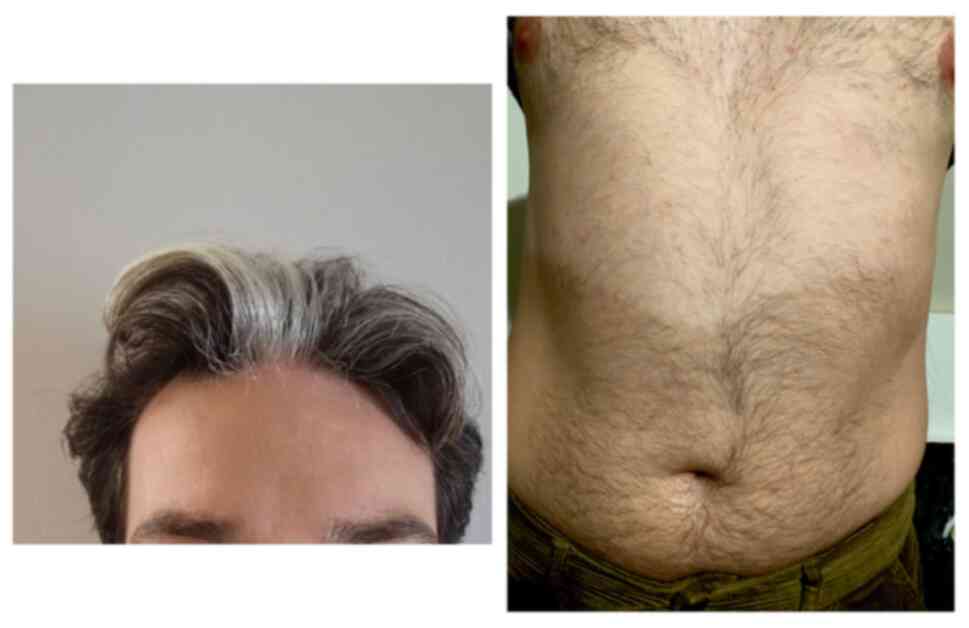
confirmed that the patient had skin pigmentation disorders,
including leukodermia, manifested by white patches of skin in the
abdominal area and pigmentary deficiencies of the hair, namely a
white lock of hair in the front-center of the head (Fig. 1). Although these features had been
evident since infancy, at the age of 33 the patient attended Access
To Genome, Genetics Laboratory (Athens, Greece) in October 2023 to
confirm the diagnosis of WS. Admission was not required and the
patient was not hospitalized.
According to the family history it was noted that
both of the patient's parents exhibited normal hearing. The
patient's brother had a hearing test as a child and the results
were normal, with no concerns regarding hearing results up to the
completion of the present study. The brother's hair started to turn
grey when he was 33 years old and a white lock of hair in the
front-center of the head was present for as long as could be
remembered. Additional information indicated that the paternal
grandmother also had patches of white hair on the head.
Furthermore, the uncle on the father's side had normal hearing and
white hair since puberty (covering the entire head); the uncle was
ischemic since the age of 25 years and developed colonic
diverticula. Finally, no other family history of sensorineural HL,
kidney problems, cancer or iris heterochromia was reported. The
clinicians, based on the clinical examination and the comprehensive
history of the patient and the patient's family, assumed that the
patient presented symptoms compatible with WS.
The patient and the family members were fully
informed regarding the nature, purpose and procedures of the study;
they received adequate information and had the opportunity to ask
questions and receive clarification. Subsequently, they provided
written consent to participate in the study.
Molecular genetic screening of the
patient
To confirm the diagnosis, genetic counseling was
recommended and molecular genetic testing of the patient was
suggested. Initially peripheral blood was collected from the
patient using standard phlebotomy procedures with EDTA
anticoagulant tubes. Genomic DNA was extracted from whole blood
using the bead-based method MagAttract HMW DNA Kit (cat. no. 67563;
Qiagen, Inc.). DNA quantity was measured fluorometrically with the
Qubit dsDNA HS Assay Kit (cat. no. Q32854; Thermo Fisher
Scientific, Inc.) and a Qubit Fluorometer. Qualified genomic DNA
was randomly fragmented by non-contact, isothermal sonochemistry
using the Covaris LE220-plus (Covaris, LLC). Fragment ends were
end-repaired, A-tailed, and ligated to sequencing adapters, IDT for
Illumina TruSeq DNA UD Indexes (cat. no. 20022370; Illumina Inc.).
Size selection was performed with a bead-based clean-up method,
AMPure XP (cat. no. A63880; Beckman Coulter, Inc.). Libraries were
PCR-amplified according to manufacturer recommendations and
enriched by hybridization-based capture using the Twist Clinical
Exome workflow with a custom Waardenburg Syndrome Panel. The custom
target set captured EDN3, EDNRB, KIT,
KITLG, MITF, PAX3, SNAI2 and
SOX10 genes, including all protein-coding exons, exon-intron
boundaries (±20 bp) and selected non-coding deep intronic variants.
Library quality was assessed by verifying insert size distribution
and absence of adapter dimers, using the Agilent 2100 Bioanalyzer
with the High Sensitivity DNA Kit (cat. no. 5067-4626; Agilent
Technologies, Inc.). Library concentrations were determined by
qPCR, with the KAPA Library Quantification Kit for Illumina (cat.
no. KK4824; Roche Diagnostics GmbH) on a real-time PCR platform.
Final libraries were normalized to 1.5-2.0 pM prior to sequencing.
Sequencing was performed on an Illumina NovaSeq™ 6000 using
paired-end 150-bp sequencing-by-synthesis chemistry with the Twist
Clinical Exome workflow. The reagent kit used was the NovaSeq 6000
SP 300-cycle kit (cat. no. 20040326; Illumina, Inc.). In-process
reference samples and control libraries were included for process
monitoring and quality control. The Waardenburg Syndrome Panel
achieved 100% of targeted bases at ≥20x coverage with a median
depth of 282x, calculated from MQ0-filtered aligned reads.
Sequencing runs met established sensitivity and specificity
thresholds. Bioinformatics and quality control were performed as
follows. Raw sequencing data (BCL files) were converted to FASTQ
using Illumina bcl2fastq v2.20 (Illumina, Inc.). Reads were aligned
to the human reference genome GRCh37/hg19 using Burrows-Wheeler
Aligner (BWA-MEM) v0.7.17 (https://github.com/lh3/bwa/releases/tag/v0.7.17).
Post-alignment processing included duplicate marking, local
realignment around indels and base quality score recalibration
performed with the Sentieon implementation of GATK algorithms.
Single-nucleotide variants and small insertions/deletions were
called per standard pipeline parameters. Variants were annotated
using VcfAnno (https://github.com/brentp/vcfanno) and Ensembl Variant
Effect Predictor (https://www.ensembl.org/info/docs/tools/vep/index.html),
referencing public databases including gnomAD (https://gnomad.broadinstitute.org/), ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD
(https://www.hgmd.cf.ac.uk/).
Sample-level quality control included assessments for
contamination, sex concordance and sample mix-ups. Coverage metrics
such as median depth and percentage of targeted bases at ≥20x were
computed across all intervals to verify adequate performance.
Exonic copy-number variants were detected using CNVkit (https://cnvkit.readthedocs.io/) in conjunction
with an in-house depth-of-coverage-based pipeline. Expected
sequencing depth for target regions was modeled using other samples
sequenced in the same batch as references, and GC-content bias was
corrected during analysis. Copy-Number Variants (CNVs) detected by
NGS were confirmed by quantitative PCR or digital PCR. Variants not
meeting stringent NGS quality metrics, such as those with low
quality scores, difficult genomic regions or complex sequence
contexts, as well as selected variants of uncertain significance,
were confirmed by bi-directional Sanger sequencing. CNVs involving
fewer than 10 exons (heterozygous) or three exons
(homozygous/hemizygous), or novel events, were confirmed
orthogonally unless previously validated by the laboratory. Variant
classification followed the American College of Medical Genetics
and Genomics/Association for Molecular Pathology (ACMG/AMP) 2015
guidelines (15). Clinical
interpretation incorporated the patient's phenotype, segregation
and clinical context, curated literature, and all sequencing and
quality control data. Findings were reviewed by a multidisciplinary
team prior to reporting.
Molecular testing of the patient revealed the
presence of a novel likely pathogenic nucleotide variant,
c.209G>A (p.Cys70Tyr), in the PAX3 gene in heterozygosity. This
missense mutation results in the substitution of guanine (G) by
adenine (A) at nucleotide position 209, leading to an amino acid
change from cysteine (Cys) to tyrosine (Tyr) at position 70 of the
PAX3 protein.
Molecular genetic screening of the
family
Based on the patient's family history and the fact
that WS can be inherited, it was considered appropriate to perform
molecular analysis to detect the aforementioned nucleotide change
in first-degree relatives. Therefore, genomic DNA was extracted
from venous blood samples obtained from the patient's mother,
father, and brother, and subsequently analyzed by PCR and Sanger
sequencing. Targeted amplification of the PAX3 gene segment
harboring the c.209G>A (p.Cys70Tyr) variant was performed by
PCR. PCR amplification was carried out using the primers PAX3_F
(5'-CGCCTTTACGCACCTTCACAA-3') and PAX3_R
(5'-GAGTCCGATGTCGAGCAGTTT-3'), designed to amplify the region
containing the variant. The primers had melting temperatures of
60-61˚C, and the PCR program RYR1-59 was applied. Each reaction was
prepared in a final volume of 50 µl, containing 10 µl of 5X PCR
buffer (final concentration 1X), 3 µl MgCl2 (25 mM
stock; final concentration 1.5 mM), 4 µl dNTP mix (2.5 mM stock;
final concentration 0.2 mM), 0.25 µl forward primer (100 µM stock;
final concentration 1.0 µM), 0.25 µl reverse primer (100 µM stock;
final concentration 1.0 µM), 0.2 µl Taq DNA polymerase (5 U/µl;
final concentration 0.04 U/µl), 5 µl genomic DNA and nuclease-free
water to reach the final volume. Thermal cycling conditions
consisted of an initial denaturation at 95˚C for 5 min, followed by
35 cycles of denaturation at 95˚C for 30 sec, annealing at 60˚C for
30 sec and extension at 72˚C for 30 sec, and then a final extension
at 72˚C for 5 min. PCR products were then held at 4˚C until further
analysis. Amplicons were evaluated by capillary electrophoresis on
the ABI 3500 Genetic Analyzer (Thermo Fisher Scientific) to confirm
product quality. Direct Sanger sequencing of the PCR products was
subsequently performed on an ABI PRISM 3130xl Genetic Analyzer
(Applied Biosystems). Variant detection and segregation analysis in
the family were carried out by aligning the obtained sequences to
the PAX3 reference sequence NM_181457.4 (https://www.ncbi.nlm.nih.gov/nuccore/NM_181457.4)
using Chromas software v2.6.6 (Technelysium Pty. Ltd.; https://technelysium.com.au/wp/chromas/).
Molecular genetic testing of the family indicated
that the patient's father and brother were heterozygous for the
same nucleotide change c.209G>A (p.Cys70Tyr) in the PAX3
gene, while the mother was found to be negative for the presence of
this mutation (Table I). The
outcome for the patient was genetic counselling only.
 | Table IMolecular testing results. |
Table I
Molecular testing results.
| Gene | Nucleotide
change | Amino acid
change | Results | Genotype | Inheritance
pattern | Clinical
interpretation |
|---|
| PAX3
(NM_181457.4) | c.209G>A | p.Cys70Tyr | Missense
variant | Heterozygous | Autosomal
dominant/recessive | Likely
pathogenic |
Discussion
Mutations in the PAX3 gene are associated
with the clinical features of WS1 and with craniofacial
deafness-hand syndrome (CDHS) following an autosomal dominant
inheritance pattern, and with WS3 following an autosomal dominant
or recessive inheritance pattern (OMIM#: 606597, 122880, 193500 and
148820) (16). There is also
evidence that a fusion between the PAX3 and FKHR
(foxo1a) genes is associated with alveolar rhabdomyosarcoma (OMIM#:
606597 and 268220). This variant, however, has not, to the best of
the authors knowledge, been reported to be associated with clinical
conditions. However, another variant at this gene position,
p.Cys70Arg, has been associated with WS, suggesting that a change
at this position adversely affects protein structure and/or
function and is potentially pathogenic (17). In general, this variant has not been
reported in the Broad Institute dataset (https://www.broadinstitute.org/datasets) (individuals
without severe disease with childhood onset). The physicochemical
difference between Cys and Tyr, as measured by the distance
‘Grantham score’, is 194. This variant has a high score and is
predicted to be harmful. This score is considered a ‘radical’
variation, indicating that Cys and Tyr have significantly different
physicochemical properties (18,19).
Amino acid conservation analysis indicates that the wild-type amino
acid, Cys70, is fully conserved in all 99 vertebrates examined,
raising the possibility that a change at this position would not be
tolerated and could adversely affect protein structure and/or
function. In addition, a previous study indicated that a similar
mutation affected a highly conserved Cys residue in the paired
region, which was predicted to be involved in DNA binding (20). In addition, this c.209G>A
(p.Cys70Tyr) nucleotide change is not recorded in the Genome
Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org) and is not reported
in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) nor has it been
described to date in patients with pax3-related diseases. This
nucleotide change is located in a gene region where several point
missense pathogenic mutations are concentrated and is expected to
have a deleterious effect on protein structure. According to the
ACMG/AMP guidelines (15), it is
classified in the category of likely pathogenic findings.
According to Jalilian et al (21), the PAX3 gene contains ~100
mutations that have been reported and only few are repeatable.
These mutations appear to be missense in 38%, minor deletions in
20%, nonsense in 15%, large deletions in 11%, splicing in 8% and
minor insertions in 8%. According to Pingault et al
(2), the majority of these
mutations are found from exon 2 (with the highest frequency) to 6,
altering the structure of PB or HD and therefore affecting the DNA
binding site. According to Baldwin et al (11), Carey et al (22) and Jalilian et al (21), a low number of mutations are
identified in exons 1, 7 and 8, comprising variations that affect
the activation site. However, no mutations have been found in exons
9 and 10. According to Hoth et al (23) and Tassabehji et al (24), 80% of WS1 cases are heterozygous
mutations. In contrast to these observations, WS3 cases have been
found to be either heterozygous or homozygous. Furthermore, partial
or total deletion or even minor mutations of PAX3 and
adjacent genes are frequently observed in WS3 cases. In addition,
Wang et al (25) and Jang
et al (26) detected a de
novo mutation in WS1 in a Chinese and a Korean population,
respectively. Furthermore, Chen et al (27) detected germline mosaicism in a rare
case of two siblings with WS1. In addition, mutations in the
PAX3 gene are also found in CDHS, which is an autosomal
dominant condition classified as an allelic variant of WS and
characterized by craniofacial abnormalities and neurosensory
deafness (28). Asher et al
(29) detected the mutation
p.Asn47Lys in exon 2 of PAX3 in a family with three affected
members.
In addition, it is worth noting that the
genotype-phenotype correlations in PAX3 are not well
documented, except for the pathogenic variants p.Asn47His in
WS3(23) and p.Asn47Lys in CDHS
(29). Baldwin et al
(11) and Tassabehji et al
(24) concluded no connection among
the type and location of the mutation and the phenotype severity.
DeStefano et al (30)
demonstrated that the presence of pigmentation abnormalities in
individuals with WS1 was more strongly associated with pathogenic
PAX3 variants that delete the HD than with missense or
deletions of pathogenic variants that include PB. Furthermore, no
genotype and phenotype association has been found for HL in WS1.
According to Milunsky et al (31) no discernible difference has been
noted in the severity between whole or partial gene deletions and
the clinical spectrum reported for small endogenous PAX3
pathogenic variants. Finally, Yang et al (32) described a case of a combined
phenotype of WS type 1 and 2, where one parent exhibited WS1 as a
result of a heterozygous pathogenic variant in PAX3 at the
intron 4 splicing site. The other parent exhibited WS2 as a result
of a heterozygous pathogenic variant in the MITF gene. As a
result, the child was heterozygous for both pathogenic variants. In
addition, the patient appeared to have significantly more
pigmentation abnormalities than both parents, indicating an
additive impact of those two gene variations.
Future research and clinical trials for WS should
focus on several promising areas, such as understanding the genetic
basis of the syndrome, developing new diagnostic methods and
exploring potential treatments. Firstly, conducting genetic
research is important to understand the genetic mechanisms behind
WS. Studies focusing on the functional characterization of novel
mutations are essential, as they involve studying the way by which
these genetic changes affect protein function and contribute to the
symptoms of WS. In addition, it is important to analyze the
relationships between specific genetic mutations and clinical
expression of the syndrome. Secondly, researchers should aim to
improve diagnostic accuracy and personalize treatment plans. The
use of NGS can aid the identification and recording of novel
etiological variants in patients, enriching international
databases. This approach aids the identification of specific
genetic mutations responsible for the disease. Finally, therapeutic
approaches should consider possible biological therapies that could
target the underlying genetic causes of WS. This includes gene
therapy and other molecular therapies that could correct or
mitigate the effects of mutations. Although assistive technologies,
such as hearing aids, cochlear implants or other hearing aids can
alleviate HL symptoms, there is currently no cure for WS. Recently,
significant progress has been made in preclinical research on
hereditary HL in animal models, including gene transfer and stem
cell replacement therapy. The review article by Huang et al
(33) highlights the current
understanding of the pathogenic mechanisms and potential biological
treatments for HL in WS, and provides strategies and directions for
the implementation of WS biological therapies and possible issues
in the future. Therefore, these areas of research are essential for
advancing the understanding of the syndrome and developing more
effective treatments.
Overall, WS exhibits several different types with
certain differences in symptoms, which may differ between patients
of the same type. The two features that are common to all types of
the syndrome are congenital sensory HL to a certain degree and
pigmentation abnormalities. The diagnosis of the syndrome can often
be difficult due to its variable symptoms and genetic variations. A
thorough medical evaluation and genetic testing are important to
determine the presence of the condition. WS has no cure and is a
difficult condition to manage, requiring a multi-scientific team to
address all complications. The present study described the case of
a 33-year-old male with clinical features and a family history
compatible with WS. The patient and first-degree relatives were
submitted to genetic testing by NGS and it was found that the
patient, father and brother were heterozygous for the novel
nucleotide change c.209G>A (p.Cys70Tyr) in the PAX3 gene,
while the mother was negative for the presence of this mutation.
The mutation is classified as a likely pathogenic finding and is
likely to be responsible for the observed pathogenicity of the
syndrome. The discovery and reporting of new mutations associated
with the syndrome contributes to the understanding of the
association between genotypes and phenotypes, which aids the
genetic counseling and diagnosis. In addition, the present study
demonstrated that NGS is a useful approach for the diagnosis of
congenital diseases and is beneficial for disease screening,
genetic diagnosis and counseling.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the European Nucleotide Archive at EMBL-EBI under accession
number (PRJEB89662) or at the following URL: https://www.ebi.ac.uk/ena/browser/view/PRJEB89662.
Authors' contributions
JZ designed the study, contributed to the analysis
and interpretation of the data, and drafted, revised and submitted
the manuscript. EP, AP, SS, OV and GKP contributed to the design of
the study and to the analysis and interpretation of the data, and
revised the manuscript critically for important intellectual
content. ES and EM participated in the study design, analyzed the
laboratory data, and provided technical expertise for the
laboratory techniques and results. JZ and EM confirm the
authenticity of all raw data. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
The present case report was prepared in accordance
with institutional policies. The patient and their family provided
consent to participate in the study.
Patient consent for publication
Written informed consent was obtained from the
patient and their family for the publication of any patient data or
associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Waardenburg PJ: A new syndrome combining
developmental anomalies of the eyelids, eyebrows and nose root with
pigmentary defects of the iris and head hair and with congenital
deafness. Am J Hum Genet. 3:195–253. 1951.PubMed/NCBI
|
|
2
|
Pingault V, Ente D, Dastot-Le Moal F,
Goossens M, Marlin S and Bondurand N: Review and update of
mutations causing Waardenburg syndrome. Hum Mutat. 31:391–406.
2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wollnik B, Tukel T, Uyguner O, Ghanbari A,
Kayserili H, Emiroglu M and Yuksel-Apak M: Homozygous and
heterozygous inheritance of PAX3 mutations causes different
types of Waardenburg syndrome. Am J Med Genet Part A. 122A:42–45.
2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zlotogora J, Lerer I, Bar-David S, Ergaz Z
and Abeliovich D: Homozygosity for Waardenburg syndrome. Am J Hum
Genet. 56:1173–1178. 1995.PubMed/NCBI
|
|
5
|
Issa S, Bondurand N, Faubert E, Poisson S,
Lecerf L, Nitschke P, Deggouj N, Loundon N, Jonard L, David A, et
al: EDNRB mutations cause Waardenburg syndrome type II in the
heterozygous state. Hum Mutat. 38:581–593. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zazo Seco C, Serrão de Castro L, van
Nierop JW, Morín M, Jhangiani S, Verver EJJ, Schraders M, Maiwald
N, Wesdorp M, Venselaar H, et al: Allelic mutations of KITLG,
encoding KIT ligand, cause asymmetric and unilateral hearing loss
and Waardenburg syndrome type 2. Am J Hum Genet. 97:647–660.
2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vona B, Schwartzbaum DA, Rodriguez AA,
Lewis SS, Toosi MB, Radhakrishnan P, Bozan N, Akın R, Doosti M,
Manju R, et al: Biallelic KITLG variants lead to a distinct
spectrum of hypomelanosis and sensorineural hearing loss. J Eur
Acad Dermatol Venereol. 36:1606–1611. 2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bondurand N, Dastot-Le Moal F, Stanchina
L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I,
Skopinski L, Reardon W, et al: Deletions at the SOX10 gene locus
cause Waardenburg syndrome types 2 and 4. Am J Hum Genet.
81:1169–1185. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Fernández RM, Núñez-Ramos R, Enguix-Riego
MV, Román-Rodríguez FJ, Galán-Gómez E, Blesa-Sánchez E, Antiñolo G,
Núñez-Núñez R and Borrego S: Waardenburg syndrome type 4: Report of
two new cases caused by SOX10 Mutations in Spain. Am J Med Genet A.
164:542–547. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang X, Zhu Y, Shen N, Peng J, Wang C, Liu
H and Lu Y: A de novo deletion mutation in SOX10 in a Chinese
family with Waardenburg syndrome type 4. Sci Rep.
7(41513)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Baldwin CT, Hoth CF, Macina RA and
Milunsky A: Mutations in PAX3 that cause Waardenburg syndrome type
I: Ten new mutations and review of the literature. Am J Med Genet.
58:115–122. 1995.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Barber TD, Barber MC, Cloutier TE and
Friedman TB: PAX3 gene structure, alternative splicing and
evolution. Gene. 237:311–319. 1999.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Boudjadi S, Chatterjee B, Sun W, Vemu P
and Barr FG: The expression and function of PAX3 in development and
disease. Gene. 666:145–157. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
National Organization for Rare Disorders
(NORD): Waardenburg Syndrome - Symptoms, Causes, Treatment.
https://rarediseases.org/rare-diseases/waardenburg-syndrome/.
Accessed June 29, 2025.
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Milunsky JM: Waardenburg Syndrome Type I.
In: GeneReviews®. Adam MP, Feldman J, Mirzaa GM, Pagon
RA, Wallace SE and Amemiya A (eds). University of Washington,
Seattle, WA, 2001.
|
|
17
|
Xiao X, Huang Y, Zhang J, Cao Y and Zhang
M: Identification of two variants in PAX3 and FBN1 in a Chinese
family with Waardenburg and Marfan syndrome via whole exome
sequencing. Funct Integr Genomics. 23(114)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Grantham R: Amino acid difference formula
to help explain protein evolution. Science. 185:862–864.
1974.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li WH, Wu CI and Luo CC: Nonrandomness of
point mutation as reflected in nucleotide substitutions in
pseudogenes and its evolutionary implications. J Mol Evol.
21:58–71. 1984.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hauswirth R, Haase B, Blatter M, Brooks
SA, Burger D, Drögemüller C, Gerber V, Henke D, Janda J, Jude R, et
al: Mutations in MITF and PAX3 cause ‘splashed white’ and other
white spotting phenotypes in horses. PLoS Genet.
8(e1002653)2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jalilian N, Tabatabaiefar MA, Farhadi M,
Bahrami T and Noori-Daloii MR: . A novel mutation in the PAX3 gene
causes Waardenburg syndrome type I in an Iranian family. Int J
Pediatr Otorhinolaryngol. 79:1736–1740. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Carey ML, Friedman TB, Asher JH Jr and
Innis JW: Septo-optic dysplasia and WS1 in the proband of a WS1
family segregating for a novel mutation in PAX3 exon 7. J Med
Genet. 35:248–250. 1998.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hoth CF, Milunsky A, Lipsky N, Sheffer R,
Clarren SK and Baldwin CT: Mutations in the paired domain of the
human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well
as Waardenburg syndrome type I (WS-I). Am J Hum Genet. 52:455–462.
1993.PubMed/NCBI
|
|
24
|
Tassabehji M, Newton VE, Liu XZ, Brady A,
Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E,
Reardon W, et al: The mutational spectrum in Waardenburg syndrome.
Hum Mol Genet. 4:2131–2137. 1995.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang J, Li S, Xiao X, Wang P, Guo X and
Zhang Q: PAX3 mutations and clinical characteristics in Chinese
patients with Waardenburg syndrome type 1. Mol Vis. 16:1146–1153.
2010.PubMed/NCBI
|
|
26
|
Jang MA, Lee T, Lee J, Cho EH and Ki CS:
Identification of a novel de novo variant in the PAX3 gene in
Waardenburg syndrome by diagnostic exome sequencing: The first
molecular diagnosis in Korea. Ann Lab Med. 35:362–365.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen K, Zhan Y, Wu X, Zong L and Jiang H:
Germinal mosaicism of PAX3 mutation caused Waardenburg syndrome
type I. Int J Pediatr Otorhinolaryngol. 104:200–204.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sommer A, Young-Wee T and Frye T:
Previously undescribed syndrome of craniofacial, hand anomalies,
and sensorineural deafness. Am J Med Genet. 15:71–77.
1983.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Asher JH Jr, Sommer A, Morell R and
Friedman TB: Missense mutation in the paired domain of PAX3 causes
craniofacial-deafness-hand syndrome. Hum Mutat. 7:30–35.
1996.PubMed/NCBI View Article : Google Scholar
|
|
30
|
DeStefano AL, Cupples LA, Arnos KS, Asher
JH Jr, Baldwin CT, Blanton S, Carey ML, da Silva EO, Friedman TB,
Greenberg J, et al: Correlation between Waardenburg syndrome
phenotype and genotype in a population of individuals with
identified PAX3 mutations. Hum Genet. 102:499–450. 1998.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Milunsky JM, Maher TA, Ito M and Milunsky
A: The value of MLPA in Waardenburg syndrome. Genet Test.
11:179–182. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yang S, Dai P, Liu X, Kang D, Zhang X,
Yang W, Zhou C, Yang S and Yuan H: Genetic and phenotypic
heterogeneity in Chinese patients with Waardenburg syndrome type
II. PLoS One. 8(e77149)2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Huang S, Song J, He C, Cai X, Yuan K, Mei
L and Feng Y: Genetic insights, disease mechanisms, and biological
therapeutics for Waardenburg syndrome. Gene Ther. 29:479–497.
2022.PubMed/NCBI View Article : Google Scholar
|