Introduction
Receptor-type tyrosine-protein phosphatase δ (PTPRD)
is a receptor type tyrosine-protein phosphatase (RPTP) that is
composed of a cell adhesion molecule-like extracellular domain and
two cytoplasmic protein tyrosine phosphatase (PTP) domains
(1,2). PTPRD is predominantly expressed in
the brain and is known to be involved in the guidance and
termination of motor neurons during embryonic development (3). PTPRD knockout mice exhibit impaired
learning and memory, also indicating that PTPRD is essential for
the organization of neural circuits (4).
It has been shown that PTPRD is frequently
mutated in various types of cancer, including lung, colon cancer
and glioblastoma (5–8). Furthermore, homozygous deletions and
epigenetic silencing of PTPRD are also found in these
cancers, indicating that PTPRD is a tumor-suppressor gene
(9–11). However, the molecular functions of
PTPRD in cancer progression are not fully understood.
The extracellular domain of PTPRD was reported to
enhance neurite outgrowth in an isoform-specific manner (12). The intracellular domain of PTPRD
interacts with cytoskeletal rearrangement factors, such as the
Liprin-α family of proteins and MIM (Missing in Metastasis, also
known as MTSS1) (13–15). These observations indicate that
PTPRD regulates the adhesion and migration of cancer cells and that
the loss of PTPRD function promotes cancer progression. In the
present study, PTPRD suppressed colon cancer cell migration and was
found to be required for appropriate cell-cell adhesion. PTPRD also
regulated cell migration in cooperation with β-catenin/TCF
signaling and its target CD44. CD44 is a receptor for hyaluronic
acid and other extracellular matrix (ECM) proteins, and is reported
to be involved in cancer invasion and metastasis (16). Furthermore, the expression levels
of PTPRD were decreased in highly invasive cancers compared to less
invasive cases, and were significantly correlated with patient
survival. These results implicate PTPRD in colon cancer cell
invasion and progression.
Materials and methods
Cell culture and transfection
DLD-1 cells were cultured in RPMI medium
supplemented with 10% fetal bovine serum (FBS). HEK293T cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing
10% FBS. RKO cells were cultured in Eagle’s minimum essential
medium (MEM) containing 10% FBS. Plasmids and siRNAs were
transfected into cells using Lipofectamine 2000 and RNAiMAX
(Invitrogen), respectively.
Plasmid construction
Myc-tagged PTPRD (corresponding to NM_002839.2) was
generated by PCR from the cDNA of HEK293T cells and cloned into
pcDNA3.1(+) (Invitrogen). For the preparation of GST-fusion
proteins, cDNA fragments were subcloned into pGEX-5X (GE
Healthcare). GST-fusion proteins were synthesized in Escherichia
coli and isolated by adsorption to glutathione-conjugated
Sepharose (GSH-Sepharose; Pharmacia). Catalytic-inactive (C1553S)
and substrate-trapping (D1521A) mutants of PTPRD were generated
using PCR mutagenesis and cloned into pcDNA3.1(+) and pGEX-5X,
respectively.
Antibodies
Mouse monoclonal antibodies to β-catenin,
Plakoglobin and E-cadherin, were obtained from BD Biosciences. The
rabbit polyclonal antibody to PTPRD was generated by immunizing
rabbits with a GST-fusion protein containing amino acids 1096–1127
of PTPRD. The antibodies were purified by affinity chromatography
using columns to which the antigens used for immunization had been
linked.
Quantitative RT-PCR
Total RNA was extracted using TRIsure (Bioline) and
reverse-transcribed into cDNA using the ReverTra Ace qPCR RT kit
(Toyobo). Real-time PCR was performed using LightCycler480 SYBR
Green I Master and a LightCycler480 Instrument (Roche). The results
were normalized with the detected value for GAPDH. Primers used in
RT-PCR were as follows: GAPDH forward, 5′-GCA CCG TCA AGG CTG AGA
AC-3′; GAPDH reverse, 5′-TGG TGA AGA CGC CAG TGG A-3′; PTPRD
forward, 5′-GCT GCT GCT CCT CAC TTT CT-3′; PTPRD reverse, 5′-CGG
GTG TTC GTG TAA ACC TT-3′.
Immunoblotting, immunoprecipitation and
GST pull-down assay
Immunoblotting, immunoprecipitation and GST
pull-down assays were performed as described previously (17).
Immunofluorescence staining
Cells were fixed with 10% formalin/PBS for 15 min
and stained overnight with each antibody at 4˚C. Primary antibodies
were diluted as follows: anti-β-catenin (1:500), anti-Plakoglobin
(1:500) and anti-E-cadherin (1:250). Staining patterns obtained
with antibodies were visualized with Alexa Fluor 488-conjugated
secondary antibodies (Molecular Probes). Cells were photographed
with a Carl Zeiss LSM510 laser scanning microscope (Carl
Zeiss).
Migration and scattering assays
Cell migration assays were performed as described
previously (18) with minor
modifications. Briefly, the underside of the filter membrane was
coated with 4 μg/ml type-I collagen (Koken, Japan), 10 μg/ml
fibronectin (Sigma-Aldrich) or 10 μg/ml laminin (Sigma-Aldrich),
respectively, and was allowed to air-dry. Transfected DLD-1 cells
(1×104 cells per well) were added to the upper
compartment of the Transwell and allowed to migrate to the
underside for 6 h. For RKO cells, 5×103 cells were
allowed to migrate for 4 h.
For scattering assays, transfected DLD-1 cells
(1.2×105) were seeded on coverslips (18 mm in diameter;
Matsunami) and cultured for 48 h. Cells were fixed with 10%
formalin/PBS for 15 min and stained with TO-PRO 3 (Molecular
Probes). For quantification of scattering, staining patterns were
photographed and analyzed by Image-J software. The scattering index
is defined as the ratio of the area of scattering nuclei divided by
that of total nuclei.
RNA interference experiments
Stealth siRNA duplexes against PTPRD were purchased
from Invitrogen. The sequences of siRNAs targeting the human PTPRD
were: PTPRD-1, 5′-ACA TCA TTC AGT TGT AGC ACA TTT C-3′; PTPRD-3,
5′-ATG ACT TGT ATG TCA CTT GAA AGG G-3′. Validated Stealth negative
control siRNA duplex with low GC content (Invitrogen) was used as a
control. DNA oligonucleotides encoding shRNA were subcloned into
pSuper-retro-puro (OligoEngine). The sequence of shRNA targeting
the human CD44 was 5′-CTG GCG CAG ATC GAT TTG AAT-3′.
Microarray analysis
Microarray datasets and clinical annotations were
obtained from the Gene Expression Omnibus (GSE5206 and GSE17537).
Signal values of each dataset were log-transformed and normalized
before analysis. The probe set IDs were converted to Ensembl gene
ID. In cases where one gene ID matched multiple probe set IDs,
probe sets with low signal intensity were excluded, and the probe
set which shows the most variance across the samples was mapped to
the gene ID. Wnt target genes were obtained from The Wnt Homepage
(http://www.stanford.edu/~rnusse/wntwindow.html) and
converted to Ensembl gene ID. Hierarchical clustering and
visualization of the results were performed using GenePattern
software, and survival analysis was performed using R software.
Results
PTPRD suppresses colon cancer cell
migration
To investigate the role of PTPRD in cell migration,
knockdown experiments were performed using two distinct small
interference RNAs (siRNAs). The expression of PTPRD was
significantly reduced in DLD-1 cells transfected with the specific
siRNAs, but not the control siRNA (Fig. 1A). When siRNA-transfected cells
were subjected to migration assays using collagen-coated Boyden
chambers, the migratory activity of these cells was significantly
increased compared to that of the control siRNAtransfected cells
(Fig. 1B). We next performed cell
scattering assays and found that the knockdown of PTPRD induced
cell scattering without growth factor stimulation (Fig. 1C and D). Consistent with these
results, ectopic expression of PTPRD resulted in the inhibition of
cell migration (Fig. 1E). Notably,
the ectopic expression of a catalytic inactive mutant of PTPRD
(PTPRD-CS) also suppressed migration to the same extent as the
wild-type, implying that phosphatase activity is dispensable for
this suppression.
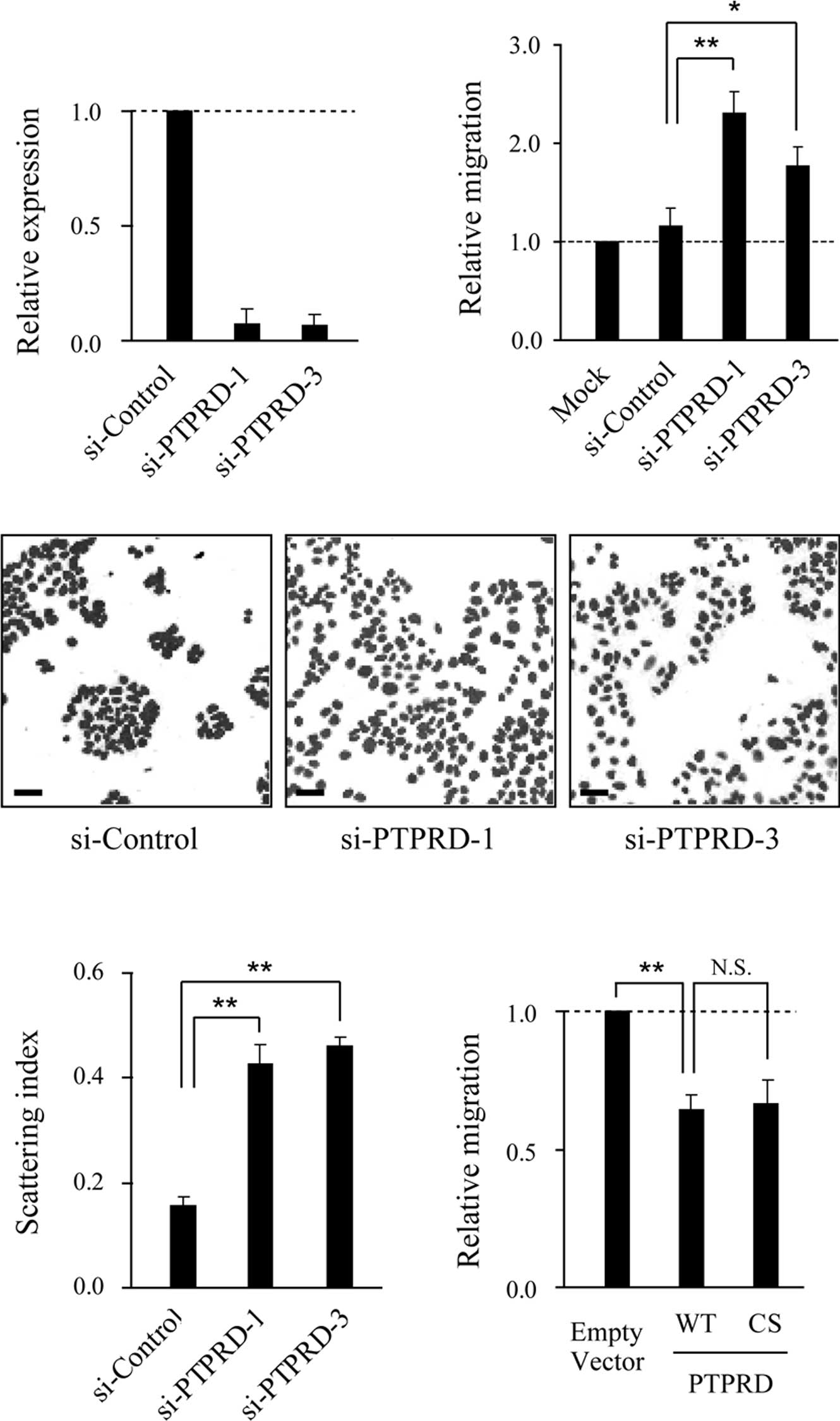 | Figure 1.PTPRD suppresses DLD-1 cell migration.
(A) Suppression of PTPRD expression by siRNAs. DLD-1 cells were
transfected with PTPRD-specific or control siRNA, respectively, and
subjected to quantitative real-time RT-PCR. Results shown represent
PTPRD mRNA expression relative to GAPDH expression. Error bars
represent the mean ± SD of three experiments in triplicate. (B)
Role of PTPRD in the migration of DLD-1 cells. Cells transfected
with the indicated siRNAs were added to the upper compartment of
the Transwell chamber and allowed to migrate to the underside of
the top chamber for 6 h. Error bars represent the mean ± SEM of at
least three independent experiments. (C) Knockdown of PTPRD induced
scattering of DLD-1 cells. Cells were transfected with
PTPRD-specific or control siRNA, respectively, and nuclear-stained
with TO-PRO 3. Scale bars, 50 μm. (D) Quantification of cell
scattering. The scattering index was calculated for the images
shown in (C) using Image J software. Bars represent the mean ± SEM
of 10 randomly selected fields. (E) DLD-1 cells were transfected
with wild-type (WT) or phosphatase inactive mutant (CS) PTPRD,
respectively, and subjected to migration assays. *p<0.05,
**p<0.01, N.S., not significant (p>0.05). |
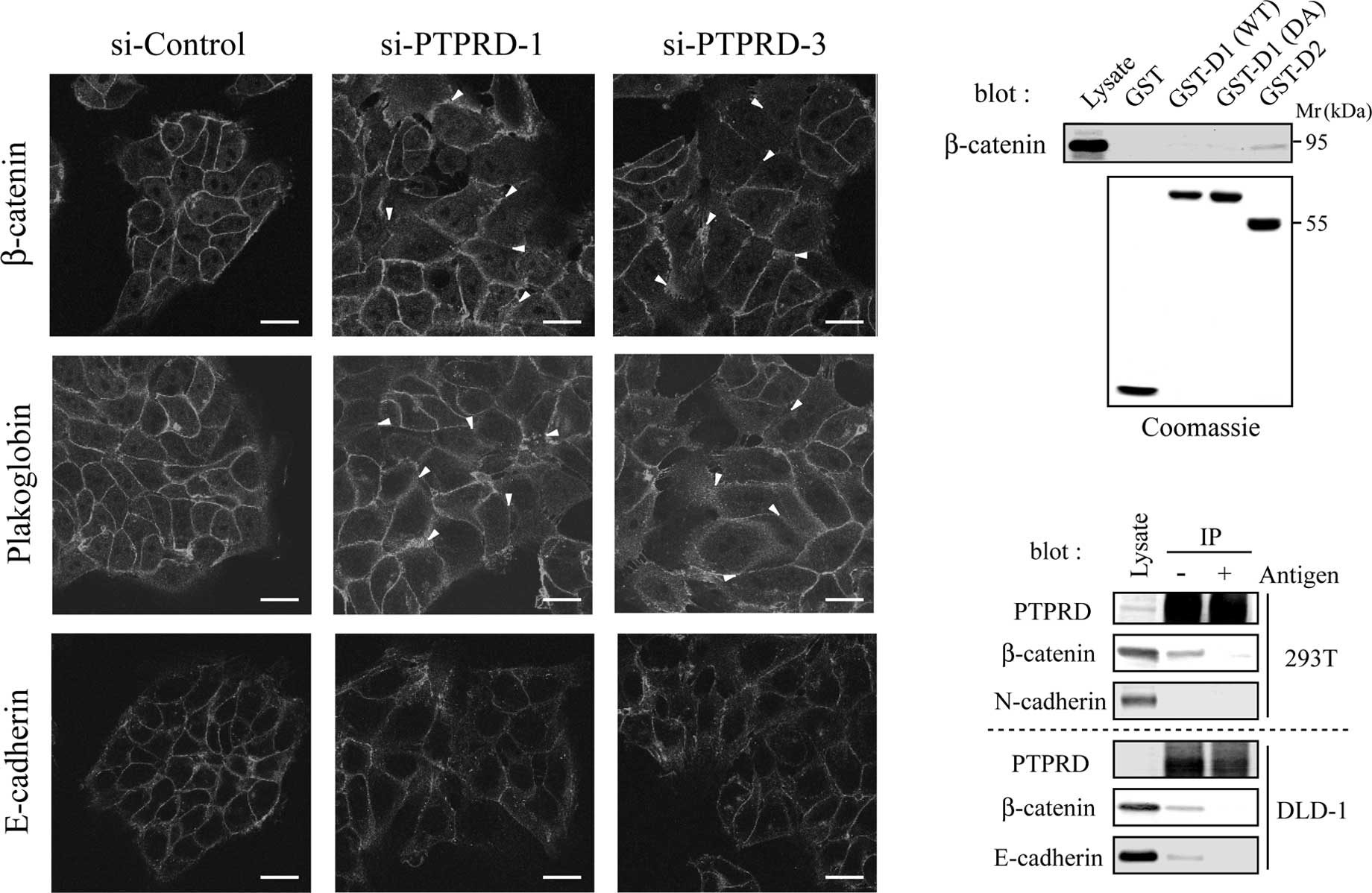
PTPRD is required for appropriate
cell-cell adhesion
It is widely accepted that the disruption of
cell-cell adhesion is a crucial step in cancer invasion and
metastasis (19). The
extracellular domain of PTPRD undergoes homophilic binding,
indicating that PTPRD plays a role in cell-cell adhesion (20). We therefore assessed the formation
of cell-cell contacts in cells transfected with PTPRD-specific
siRNAs by immunostaining for the adhesion molecules, β-catenin,
Plakoglobin and E-cadherin. These adhesion molecules clearly
accumulated at the cell-cell contacts in cells transfected with
control siRNA, whereas they showed diffused localization in cells
transfected with PTPRD-specific siRNAs (Fig. 2A, arrowheads). These results
suggest that PTPRD is required for appropriate cell-cell
adhesion.
We next investigated whether PTPRD directly
interacts with these adhesion molecules. It is known that the
membraneproximal D1 domain is catalytically active, while distal D2
domain and substrate-trapping DA mutant are inactive (1,2). We
constructed GST-fusion proteins containing one of the two PTP
domains (D1 and D2) or a substrate trapping mutant of D1 (DA),
respectively. When cell lysates were subjected to pull-down assays
with these GST-fusion proteins, β-catenin was found to interact
with D2, but not D1, DA or GST alone (Fig. 2B). To confirm this result, in
vivo immunoprecipitation experiments were performed with
HEK293T and DLD-1 cells using anti-PTPRD antibody. Both β-catenin
and E-cadherin co-precipitated with PTPRD, whereas N-cadherin did
not (Fig. 2C). In these
experiments, co-precipitations of β-catenin and E-cadherin were
inhibited by pre-incubation of the antibody with the antigen used
for immunization. These results suggest that PTPRD directly
regulates the function of the β-catenin/E-cadherin complex.
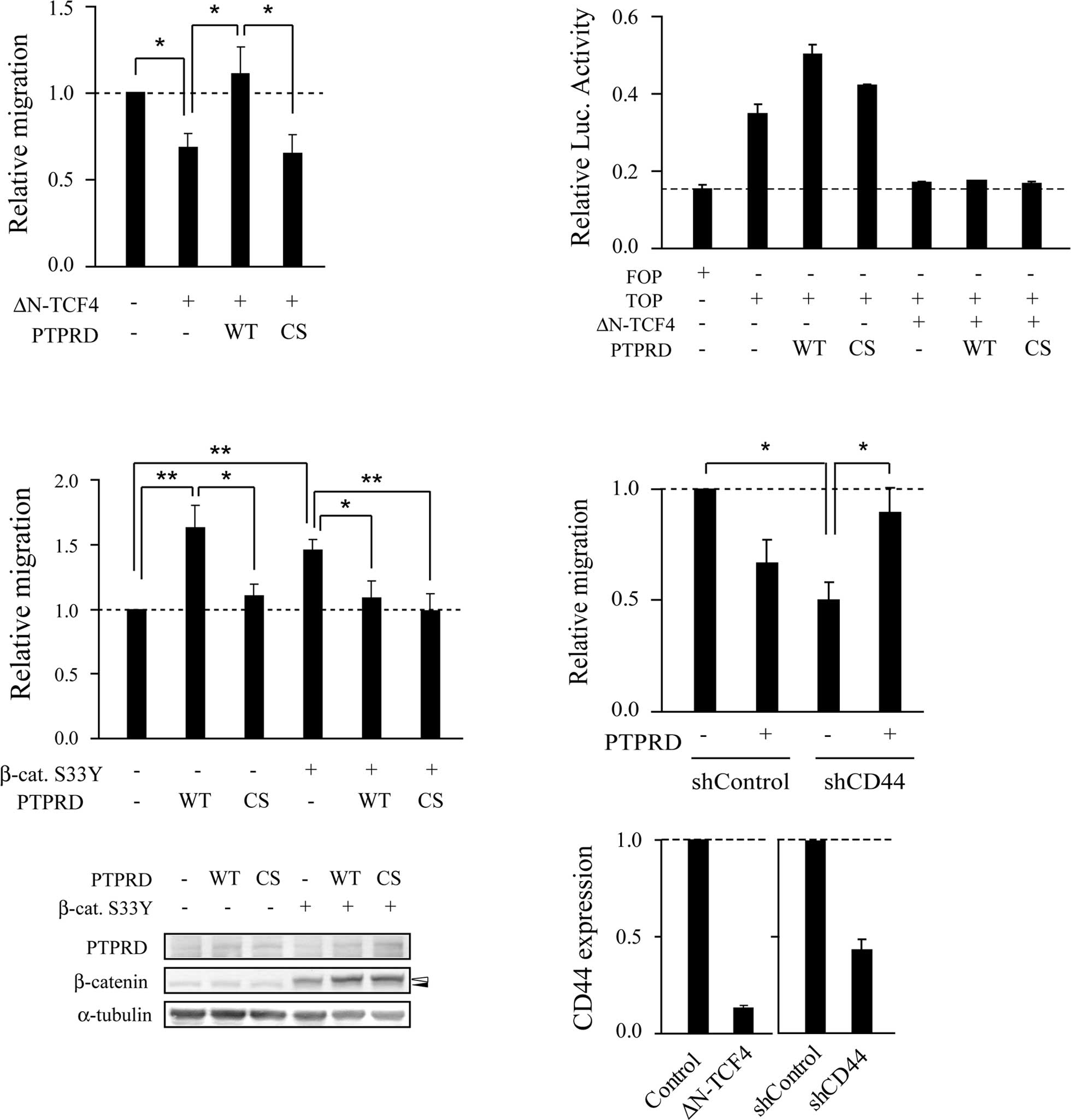
PTPRD regulates cell migration in
cooperation with β-catenin/TCF signaling
DLD-1 cells harbor biallelic nonsense mutations of
APC and express truncated mutant APC. Truncated mutant APC is
capable of enhancing cell migration through activation of both
β-catenin/TCF and Rac/Cdc42 signaling (18,21–24).
Consistent with previous reports (22–24),
ectopic expression of ΔN-TCF4, a dominant-negative form of TCF4,
suppressed cell migration (Fig.
3A). The migratory activity of cells transfected with ΔN-TCF4
along with wild-type PTPRD was unexpectedly significantly higher
than that of cells transfected with ΔN-TCF4 alone. By contrast,
PTPRD-CS, a catalytic inactive mutant, did not show this effect.
Thus, it appears that PTPRD stimulates cell migration in a
phosphatase activity-dependent manner in the absence of Wnt
signaling. To rule out the possibility that PTPRD overcomes or
inhibits the dominant-negative effect of ΔN-TCF4, luciferase assays
were performed using a TCF-responsive reporter. PTPRD failed to
increase its reporter activity when co-expressed with ΔN-TCF4
(Fig. 3B). We next performed
migration assays using RKO cells, which contain intact APC and
β-catenin. RKO cells transfected with PTPRD showed increased
motility in a phosphatase activity-dependent manner compared to the
control cells (Fig. 3C). In
contrast, when β-catenin-S33Y, a constitutively activated form of
β-catenin, was co-expressed, PTPRD suppressed cell migration in a
phosphatase activity-independent manner, as observed with DLD-1
cells in Fig. 1E. Taken together,
these results suggest that the effect of PTPRD on cell migration
depends on the status of β-catenin/TCF signaling.
We therefore attempted to identify Wnt target genes
that are involved in the regulation of PTPRD-dependent migration.
Knockdown experiments were performed using shRNAs targeting known
Wnt target genes. Similar to cells transfected with PTPRD, cells
transfected with a shRNA targeting CD44 (shCD44) showed decreased
motility compared to control cells (Fig. 3D). However, cells transfected with
PTPRD along with shCD44 showed increased migratory activity
compared to cells transfected with shCD44 alone. These observations
suggest that CD44 is involved in PTPRD-dependent migration.
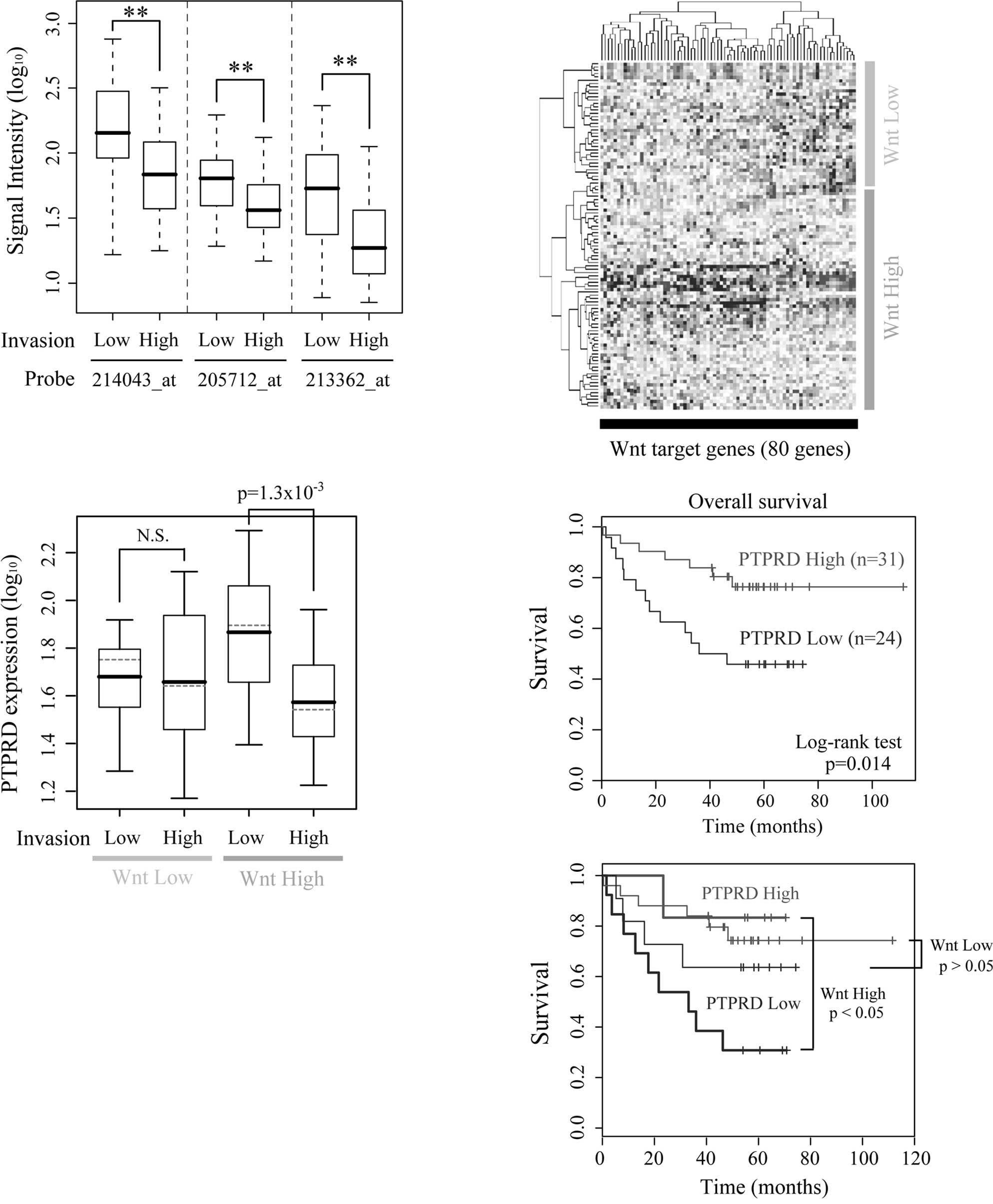
The expression levels of PTPRD are low in
highly invasive cancers and correlate with patient survival
To investigate the clinical importance of PTPRD,
gene expression profiles of human colon cancers were obtained from
a public microarray database. The expression levels of PTPRD were
significantly lower in the highly invasive cancers (T stage 3 or 4)
than in the less invasive cancers (Fig. 4A). There were no significant
differences in either lymph node and distal metastasis (data not
shown). In most colon cancers, APC or β-catenin is mutated and
β-catenin/TCF signaling is constitutively activated (25,26).
However, the levels of β-catenin/TCF signaling differ among cancers
according to the type and/or the combinations of mutations
(27). We therefore divided the
dataset into two subgroups based on the expression levels of known
Wnt target genes and termed them the ‘Wnt High group’ and ‘Wnt Low
group’, respectively (Fig. 4B).
Samples from normal colons were classified as the Wnt Low group. An
association of β-catenin/TCF signaling and PTPRD expression was
noted in the Wnt High group, but not in the Wnt Low group (Fig. 4C). We also performed Cox’s
regression analysis and found that both overall survival and
disease-free survival were correlated with the expression level of
PTPRD (p=0.008 and p=0.0025, respectively; Fig. 4D, upper panel). Furthermore, a
significant correlation between PTPRD expression and survival was
observed only in the Wnt High group, but not in the Wnt Low group
(p=0.036 and p=0.22, respectively; Fig. 4D, lower panel). Taken together,
these results suggest the possibility that the down-regulated
expression of PTPRD contributes to the invasive properties and poor
patient outcome in human colon cancers in coordination with
β-catenin/TCF signaling.
Discussion
In the present study, PTPRD suppressed cancer cell
migration in cooperation with β-catenin/TCF signaling. Furthermore,
CD44, a target of Wnt signaling (28,29),
was required for PTPRD-mediated regulation of cell migration. When
β-catenin/TCF signaling was activated, thereby enhancing CD44
expression, PTPRD suppressed the migratory activity of the cancer
cells. It is well known that CD44 interacts with ERM proteins and
regulates actin polymerization and cell migration (16). Moreover, aberrant expression and
splicing of CD44 were found to be correlated with invasion and
patient outcome in colon cancer. Our observation suggests that
PTPRD and CD44 regulate cell migration and progression in
cooperation. Further study is required to clarify the molecular
mechanisms underlying this cooperation. In addition to migration
and invasion, CD44 was recently reported to be a marker for cancer
stem cells in breast and colon cancer (30,31).
Notably, both CD44 and PTPRD were found to be enriched in
Lgr5-positive mouse crypt stem cells (32). These observations suggest that
PTPRD and CD44 function cooperatively in stem cell maintenance. The
role of PTPRD in cancer stem cells is currently under investigation
in our laboratory.
We found that reduced expression of PTPRD was
correlated with invasive status and poor survival in colon cancer
patients. Furthermore, significant correlations were observed only
in the patients where Wnt signaling was highly activated. This
observation suggests that the effect of PTPRD on cell migration
depends on the status of β-catenin/TCF signaling. Activation of
β-catenin/TCF signaling is also observed in glioblastoma and
melanoma (33,34), in which homozygous deletions and
epigenetic silencing of PTPRD are found. Hence, investigation of
the association of PTPRD and β-catenin/TCF signaling in these
cancers is warranted.
Our migration assays revealed that PTPRD regulates
cell migration in a catalytic activity-independent manner. In this
regard, it is interesting that PTPRD has been reported to interact
with the Liprin-α family of proteins and MIM through its catalytic
inactive D2 domain. Liprin-α is a multi-domain scaffolding protein
that localizes RPTP to cell focal adhesions and regulates their
disassembly. MIM is an actin binding protein that induces cell
shape changes, such as the formation of lamellipodia- and
filopodia-like structures. The expression level of MIM is
negatively correlated with the metastatic activities of various
cancer types, indicating its suppressive role in cell migration and
invasion. These observations suggest that PTPRD regulates cell
migration in conjunction with these cytoskeletal rearrangement
factors via its catalytic inactive D2 domain.
We also investigated the role of PTPRD in cell-cell
adhesion and showed that PTPRD is required for the formation of
appropriate cell-cell contacts. In addition, we found that PTPRD
interacts with cell-adhesion molecules, such as β-catenin and
E-cadherin, through its D2 domain. It is known that LAR and PTPRS,
homologs of PTPRD, dephosphorylate β-catenin and thereby stabilize
cell adhesion and inhibit cell migration and neurite outgrowth
(35,36). It is therefore possible that
β-catenin is a substrate of PTPRD, and that the suppression of
PTPRD leads to disruption of the β-catenin/E-cadherin complex and
cell-cell adhesion.
In the absence of Wnt signaling, PTPRD enhanced cell
migration in a phosphatase activity-dependent manner. Some protein
tyrosine phosphatases are known to dephosphorylate and activate the
Src family of kinases (37,38).
Thus, it is also possible that PTPRD activates the Src family of
kinases and thereby increases the migratory activity of cancer
cells. Furthermore, it has recently been reported that PTPRD
dephosphorylates STAT3 and inhibits cell proliferation and
transformation (11). In general,
tyrosine phosphatases dephosphorylate a wide-range of substrates.
Therefore, further efforts to search for novel substrates are
required for the better understanding of the function of PTPRD in
cancer progression.
Acknowledgements
The authors thank H. Clevers for
providing the FOP/TOP reporter plasmids. This study was supported
by Innovative Cell Biology by Innovative Technology, Grants-in-Aid
for Scientific Research on Innovative Areas, and in part by the
Global COE program (Integrative Life Science Based in the Study of
Biosignaling Mechanisms), MEXT, Japan.
References
|
1.
|
Tiganis T and Bennett A: Protein tyrosine
phosphatase function: the substrate perspective. Biochem J.
402:1–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ensslen-Craig S and Brady-Kalnay S:
Receptor protein tyrosine phosphatases regulate neural development
and axon guidance. Dev Biol. 275:12–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Stepanek L, Stoker A, Stoeckli E and Bixby
J: Receptor tyrosine phosphatases guide vertebrate motor axons
during development. J Neurosci. 25:3813–3823. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Uetani N, Kato K, Ogura H, et al: Impaired
learning with enhanced hippocampal long-term potentiation in
PTPdeltadeficient mice. EMBO J. 19:2775–2785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Sjöblom T, Jones S, Wood L, et al: The
consensus coding sequences of human breast and colorectal cancers.
Science. 314:268–274. 2006.
|
|
6.
|
Weir B, Woo M, Getz G, et al:
Characterizing the cancer genome in lung adenocarcinoma. Nature.
450:893–898. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Network CGAR: Comprehensive genomic
characterization defines human glioblastoma genes and core
pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bignell G, Greenman C, Davies H, et al:
Signatures of mutation and selection in the cancer genome. Nature.
463:893–898. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Sato M, Takahashi K, Nagayama K, et al:
Identification of chromosome arm 9p as the most frequent target of
homozygous deletions in lung cancer. Genes Chromosomes Cancer.
44:405–414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Solomon D, Kim J, Cronin J, et al:
Mutational inactivation of PTPRD in glioblastoma multiforme and
malignant melanoma. Cancer Res. 68:10300–10306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Veeriah S, Brennan C, Meng S, et al: The
tyrosine phosphatase PTPRD is a tumor suppressor that is frequently
inactivated and mutated in glioblastoma and other human cancers.
Proc Natl Acad Sci USA. 106:9435–9440. 2009. View Article : Google Scholar
|
|
12.
|
Gonzalez-Brito M and Bixby J: Differential
activities in adhesion and neurite growth of fibronectin type III
repeats in the PTP-delta extracellular domain. Int J Dev Neurosci.
24:425–429. 2006. View Article : Google Scholar
|
|
13.
|
Serra-Pagès C, Medley Q, Tang M, Hart A
and Streuli M: Liprins, a family of LAR transmembrane
protein-tyrosine phosphataseinteracting proteins. J Biol Chem.
273:15611–15620. 1998.PubMed/NCBI
|
|
14.
|
Woodings J, Sharp S and Machesky L: MIM-B,
a putative metastasis suppressor protein, binds to actin and to
protein tyrosine phosphatase delta. Biochem J. 371:463–471. 2003.
View Article : Google Scholar
|
|
15.
|
Gonzalez-Quevedo R, Shoffer M, Horng L and
Oro A: Receptor tyrosine phosphatase-dependent cytoskeletal
remodeling by the hedgehog-responsive gene MIM/BEG4. J Cell Biol.
168:453–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ponta H, Sherman L and Herrlich P: CD44:
from adhesion molecules to signalling regulators. Nat Rev Mol Cell
Biol. 4:33–45. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nakamura T, Hayashi T, Nasu-Nishimura Y,
et al: PX-RICS mediates ER-to-Golgi transport of the
N-cadherin/beta-catenin complex. Genes Dev. 22:1244–1256. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kawasaki Y, Sato R and Akiyama T: Mutated
APC and Asef are involved in the migration of colorectal tumour
cells. Nat Cell Biol. 5:211–215. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jeanes A, Gottardi C and Yap A: Cadherins
and cancer: how does cadherin dysfunction promote tumor
progression? Oncogene. 27:6920–6929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Wang J and Bixby J: Receptor tyrosine
phosphatase-delta is a homophilic, neurite-promoting cell adhesion
molecular for CNS neurons. Mol Cell Neurosci. 14:370–384. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kawasaki Y, Sagara M, Shibata Y, Shirouzu
M, Yokoyama S and Akiyama T: Identification and characterization of
Asef2, a guanine-nucleotide exchange factor specific for Rac1 and
Cdc42. Oncogene. 26:7620–7267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gilles C, Polette M, Mestdagt M, et al:
Transactivation of vimentin by beta-catenin in human breast cancer
cells. Cancer Res. 63:2658–2664. 2003.PubMed/NCBI
|
|
23.
|
Gavert N, Conacci-Sorrell M, Gast D, et
al: L1, a novel target of beta-catenin signaling, transforms cells
and is expressed at the invasive front of colon cancers. J Cell
Biol. 168:633–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Vignjevic D, Schoumacher M, Gavert N, et
al: Fascin, a novel target of beta-catenin-TCF signaling, is
expressed at the invasive front of human colon cancer. Cancer Res.
67:6844–6853. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Morin P, Sparks A, Korinek V, et al:
Activation of beta-catenin-Tcf signaling in colon cancer by
mutations in beta-catenin or APC. Science. 275:1787–1790. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Sparks A, Morin P, Vogelstein B and
Kinzler K: Mutational analysis of the APC/beta-catenin/Tcf pathway
in colorectal cancer. Cancer Res. 58:1130–1134. 1998.PubMed/NCBI
|
|
27.
|
Fodde R and Brabletz T: Wnt/beta-catenin
signaling in cancer stemness and malignant behavior. Curr Opin Cell
Biol. 19:150–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wielenga V, Smits R, Korinek V, et al:
Expression of CD44 in Apc and Tcf mutant mice implies regulation by
the WNT pathway. Am J Pathol. 154:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Boon E, van der Neut R, van de Wetering M,
Clevers H and Pals S: Wnt signaling regulates expression of the
receptor tyrosine kinase met in colorectal cancer. Cancer Res.
62:5126–5128. 2002.PubMed/NCBI
|
|
30.
|
Al-Hajj M, Wicha M, Benito-Hernandez A,
Morrison S and Clarke M: Prospective identification of tumorigenic
breast cancer cells. Proc Natl Acad Sci USA. 100:3983–3988. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Du L, Wang H, He L, et al: CD44 is of
functional importance for colorectal cancer stem cells. Clin Cancer
Res. 14:6751–6760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Van der Flier L, van Gijn M, Hatzis P, et
al: Transcription factor achaete scute-like 2 controls intestinal
stem cell fate. Cell. 136:903–912. 2009.PubMed/NCBI
|
|
33.
|
Zheng H, Ying H, Wiedemeyer R, et al:
PLAGL2 regulates Wnt signaling to impede differentiation in neural
stem cells and gliomas. Cancer Cell. 17:497–509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
O’Connell M and Weeraratna A: Hear the Wnt
Ror: how melanoma cells adjust to changes in Wnt. Pigment Cell
Melanoma Res. 22:724–739. 2009.PubMed/NCBI
|
|
35.
|
Müller T, Choidas A, Reichmann E and
Ullrich A: Phosphorylation and free pool of beta-catenin are
regulated by tyrosine kinases and tyrosine phosphatases during
epithelial cell migration. J Biol Chem. 274:10173–10183.
1999.PubMed/NCBI
|
|
36.
|
Siu R, Fladd C and Rotin D: N-cadherin is
an in vivo substrate for protein tyrosine phosphatase sigma
(PTPsigma) and participates in PTPsigma-mediated inhibition of axon
growth. Mol Cell Biol. 27:208–219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Harder K, Moller N, Peacock J and Jirik F:
Protein-tyrosine phosphatase alpha regulates Src family kinases and
alters cellsubstratum adhesion. J Biol Chem. 273:31890–31900. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Zhu S, Bjorge J and Fujita D: PTP1B
contributes to the oncogenic properties of colon cancer cells
through Src activation. Cancer Res. 67:10129–10137. 2007.
View Article : Google Scholar : PubMed/NCBI
|