Introduction
Malignant mesothelioma (MM) of the serosal membranes
of body cavities is a particularly aggressive cancer characterized
by rapid progression, late metastases and poor prognosis (1). Although surgery, radiotherapy,
chemotherapy and/or their combinations have been used as
therapeutic modalities, median patient survival is 8–18 months
(2). Cisplatin has been used in
clinical MM therapy, and its chemotherapeutic effect as a single
agent as well as in combination with other chemotherapeutic agents
has been previously examined (3,4).
However, most patients relapsed within 1 year after starting
treatment. Therefore, new therapeutic approaches are required for
MM patients.
Among the different types of cell-cell interactions
in mammalian cells, gap junctional intercellular communication
(GJIC) is considered to be the only route allowing free direct
transfer of ions and hydrophilic molecules of up to 1,000–1,500 Da
in size between cells, thereby maintaining electrical and metabolic
cell homeostasis (5). The gap
junction is made up of juxtaposed transmembrane hemichannels
(connexons) provided by adjacent cells, and each connexon consists
of six individual transmembrane proteins called connexin (Cx)
(6). In general, it is well known
that the Cx gene acts as a tumor-suppressor gene by maintaining
homeostatic control in multicellular organisms via GJIC. Moreover,
many transfection studies using Cx cDNAs revealed that Cx is a
tumor suppressor in cells originating from tissues in which they
are normally expressed (7). In
line with this, we recently reported that Cx43 abrogates various
malignant phenotypes in MM cells, such as chemoresistance (8).
Protease inhibitors are a class of well-established
cancer chemopreventive agents due to their strong anticarcinogenic
activity in vivo and in vitro in cancer model systems
(9). The most predominant protease
inhibitor in soybeans is the Bowman-Birk inhibitor (BBI) (9). BBI, a 71-amino acid protein (8 kDa)
and a serine protease inhibitor with both trypsin and chymotrypsin
inhibitory activities, was found to be a valid suppressor of
carcinogenesis in a human phase IIa clinical trial (10). Although BBI has a broad spectrum of
cancer-protective activities (10–12),
knowledge of the exact mechanism(s) by which BBI exerts its
anticarcinogenic effects remains limited. In our previous studies,
we demonstrated that the induction of Cx43 by BBI contributes to
the negative growth control of tumor cells in vivo as well
as in vitro (13,14). Overall, it appears that BBI
improves chemoresistance in MM cells via the induction of Cx43.
In this context, we evaluated whether BBI enhances
cisplatin-induced cytotoxicity in MM cells. The H28 human MM cell
line was chosen to evaluate the inhibitory effect of BBI on MM cell
growth, due to its resistance to cisplatin, a representative agent
used to clinically treat MM.
Materials and methods
Chemicals
All culture chemicals and BBI were purchased from
Gibco BRL (Tokyo, Japan) and Sigma (St. Louis, MO, USA), unless
otherwise indicated. SU6656 (an Src inhibitor) was purchased from
Calbiochem-Novabiochem (La Jolla, CA, USA). Anti-Cx43 antibody was
from Zymed Laboratories (San Francisco, CA, USA). Other antibodies
were purchased from Wako Pure Chemicals (Osaka, Japan) and BD
Transduction Laboratories (Lexington, KY, USA).
Cell culture and treatment
Human mesothelioma H28 cell line was supplied by
ATCC, and the cells were cultured in RPMI-1640 supplemented with
10% fetal bovine serum (FBS), 0.01 M HEPES buffer solution, 1 mM
sodium pyruvate and 4.5 g/l glucose in a 5% CO2
atmosphere at 37°C in a humidified incubator. BBI was dissolved in
saline, and the cells were treated with BBI (200 and 400 μg/ml) or
vehicle (saline) for indicated periods. Treatment conditions
involving the other agents were performed as described in each
figure legend.
Cell growth assay
Cells (2×104) were seeded on a 96-well
culture plate with the culture medium, and after overnight culture,
the cells were treated with each agent. Following this, cell growth
was determined with a cell proliferation assay kit using WST-1
reagent (Roche Japan, Tokyo, Japan).
Apoptosis analysis
Cells were trypsinization, washed with PBS,
resuspended in 70% ethanol and maintained at 4°C for at least 30
min. Before analysis, the cells were washed again with PBS and
resuspended and incubated for 30 min in PBS containing 0.05 mg/ml
propidium iodide, 1 mM EDTA, 0.1% Triton X-100 and 1 mg/ml RNase A.
The suspension was then passed through a nylon mesh filter and
analyzed on a Becton Dickinson FACScan.
Measurement of GJIC
For measurement of GJIC, we used a
scrape-loading/dye transfer method with some modification (15). H28 cells on 35-mm dishes were
rinsed several times with PBS. The center of the dish was scraped
by a surgical blade, and 2 ml of 0.05% Lucifer yellow CH (LY) in
PBS was added to the dishes after scraping. LY is a small molecule
(457 Da) that freely moves through gap junctions from loaded cells
to neighboring ones. Five minutes after the dye treatment, the
cells were rinsed several times with PBS to remove excess dye. The
intensity of LY transfer was observed with an Olympus inverse
microscope equipped with appropriate filters (Olympus, Tokyo,
Japan), five points were photographed per dish and the cell layers
into which LY had spread were counted.
Cx43 antisense phosphorothioate
oligodeoxynucleotide treatment
Cx43 antisense phosphorothioate oligodeoxynucleotide
(AS-ODN) (5′-CTCCAGTCACCCATGTTG-3′) was purchased from
Sigma-Genosys (Hokkaido, Japan). The sequence encompassed the start
codon of rat Cx43 mRNA. As a control for the non-specific effects
of oligonucleotide treatment, the corresponding sense
phosphorothioate ODN (S-ODN) (5′-CAACATGGGTGACTGGAG-3′) was also
obtained from Sigma Genosys. The cells were exposed to 2 μM AS-ODN
or S-ODN and added to the culture medium at 2-day intervals. Under
these conditions, the expression of Cx43 protein was almost
diminished in the cells treated with AS-ODN, while S-ODN treatment
at the equivalent dose did not affect the protein level.
Isolation of total RNA and real-time
PCR
Total RNA was isolated by using the SV Total RNA
isolation system (Promega, Madison, WI, USA), and cDNA was
synthesized as previously described (8). Real-time PCR was performed by using
ABI Prism 7000 sequence detection system (Applied Biosystems Japan
Ltd., Tokyo, Japan) and SYBR Premix Ex Taq™ (Takara Bio Inc.,
Shiga, Japan) according to the manufacturer’s instructions. The
primers used were as follows: glyceraldehyde-3-phosphate
dehydrogenase (GAPDH), accession no. (BC023632), sense (nucleotides
737–756), antisense (nucleotides 916-897); Cx43, accession no.
(NM_000165), sense (nucleotides 257–276) and antisense (nucleotides
433-414).
Immunoblot analysis
Immunoblotting was performed as previously described
(14). Briefly, the cell lysate
was prepared in cell lysis/extraction reagent (Sigma), and 10 μg
total protein extract from each sample was loaded onto 8%
SDS-polyacrylamide gel. After electrophoresis, proteins were
transferred to nitrocellulose membranes. The blots were incubated
with each antibody. Each immunoreactive band was detected using the
ECL system (Amersham) and a cooled CCD camera-linked Cool Saver
system (Atto, Osaka Japan). Molecular sizing was carried out using
Rainbow MW marker (Amersham). Protein concentrations were
determined using the DC protein assay system (Bio-Rad, Hercules,
CA, USA).
Assay for proteasome activity
H28 cells were seeded on a 12-well plate
(5×104 cells/well) overnight. These cells were then
treated with the specific concentration of BBI for 24 or 72 h,
followed by the addition of 20 μM of flurogenic peptide substrate
Suc-Leu-Leu-Val-Tyr-AMC (for chymotrypsin-like activity) at 37°C
for 2 h. Afterwards, 100 μl of the cell medium was collected and
diluted with 1X PBS to 400 μl. Measurement of free AMC groups was
performed as previously described (16).
Statistical analysis
Data were analyzed by one-way ANOVA followed by the
Student’s t-test, Dunnett’s multiple-range test or Tukey-Kramer
test. P≤0.05 was considered statistically significant.
Results
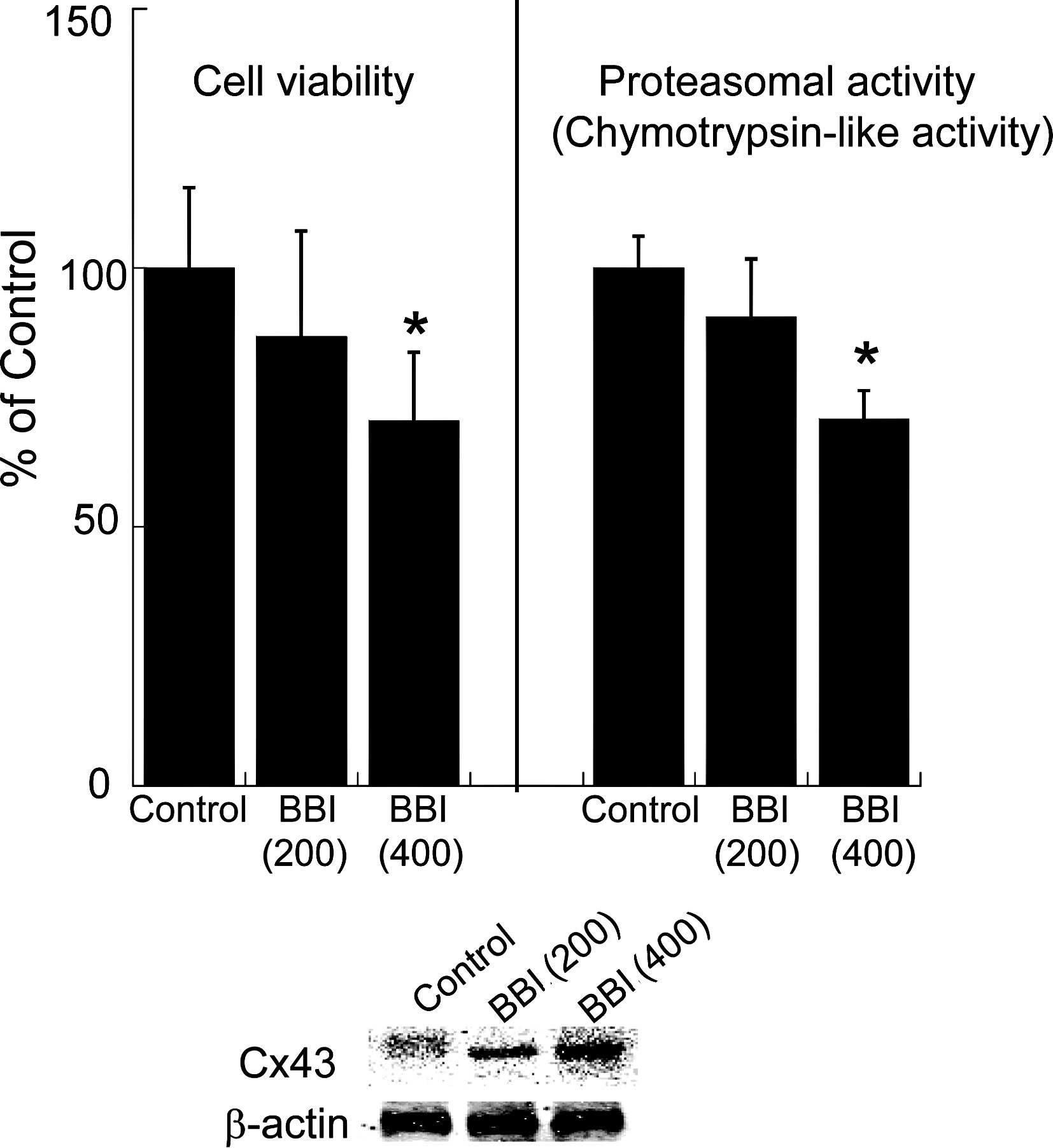
Effect of BBI on cell growth, proteasomal
activity and C43 expression in H28 cells
To evaluate the effect of BBI on cell growth,
proteasomal activity and Cx43 expression level, dose-dependent
changes in cell viability, chymotrypsin-like activity involved in
the proteasome and the protein level of Cx43 after BBI exposure for
each time period were examined. Since the elevation of Cx43 by BBI
partly depends on the inhibition of chymotrypsin-like activity in
the proteasome (14), we assessed
the activity in the proteasome to confirm the effect of BBI. As
shown in Fig. 1, BBI decreased
cell viability and suppressed proteasomal chymotrypsin-like
activity in a dose-dependent manner, whereas the expression level
of Cx43 protein was dose-dependently elevated by BBI treatment.
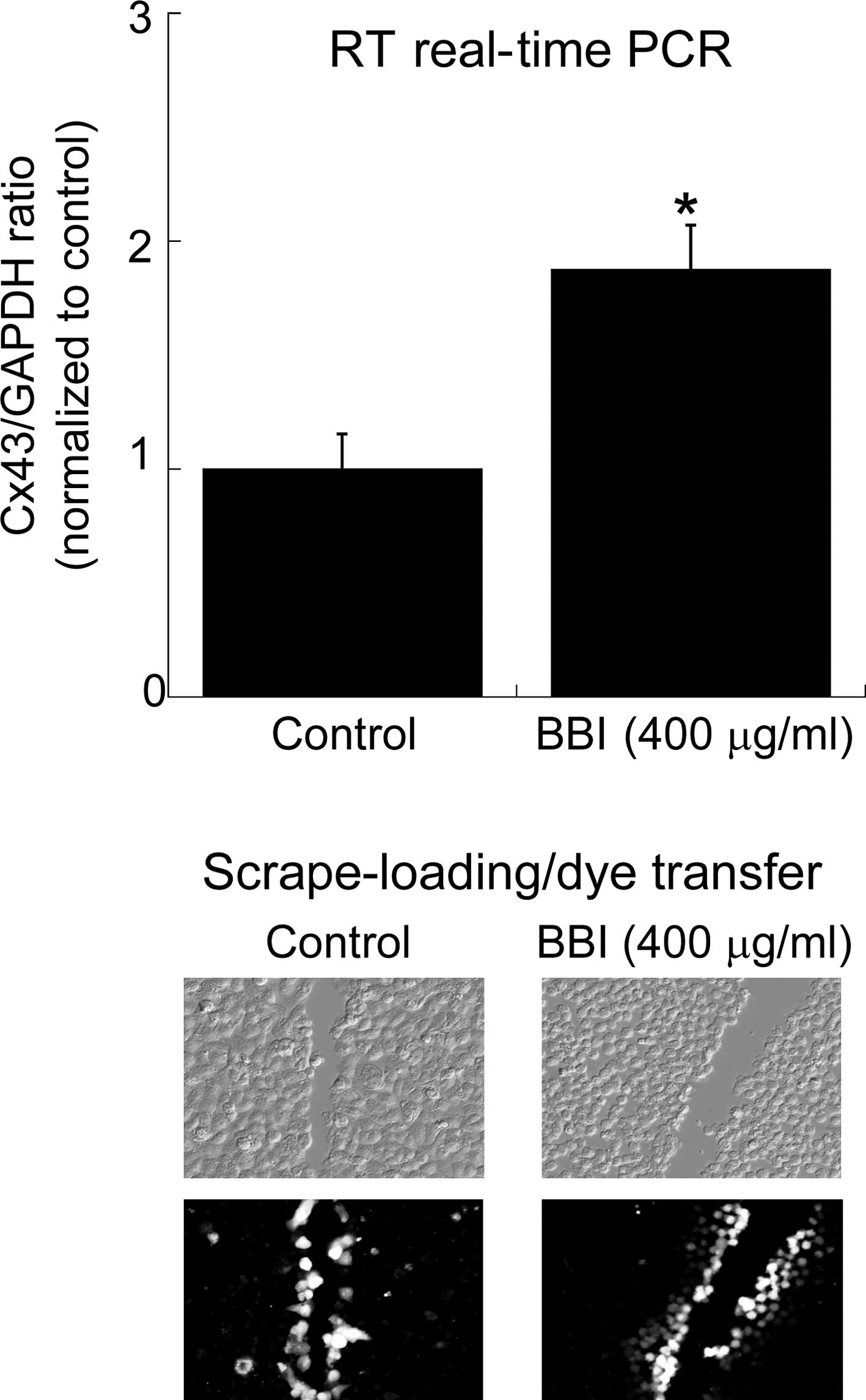
Next, the effect of BBI on the Cx43 mRNA level and GJIC in H28
cells was investigated (Fig. 2).
BBI treatment up-regulated the Cx43 mRNA level and in parallel
caused the restoration of GJIC estimated by the scrape-loading
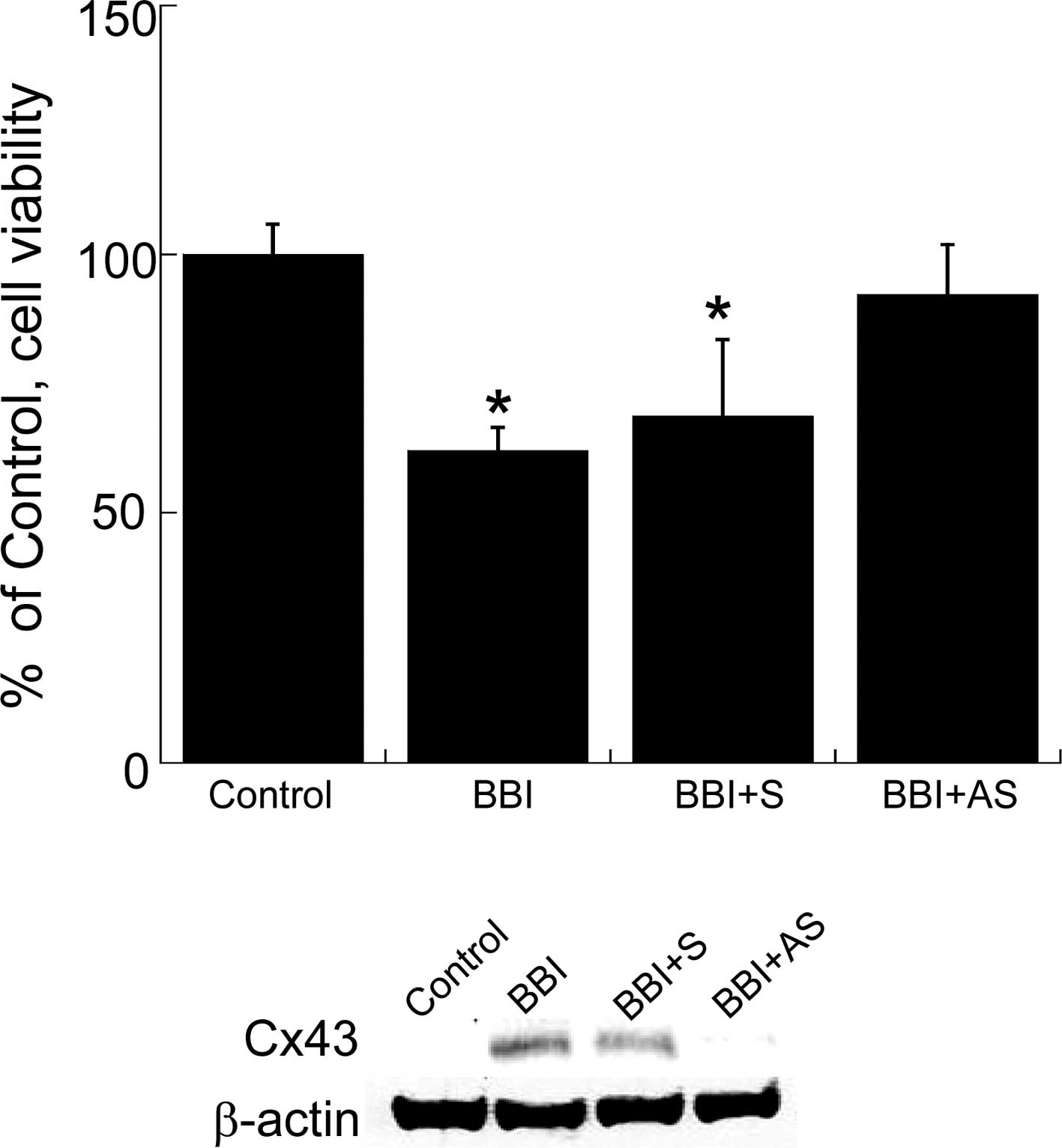
assay. Finally, to confirm the contribution of the BBI-mediated
elevation of Cx43 to the negative growth effect in H28 cells, we
ascertained whether the knockdown of Cx43 by AS-ODN abrogates the
BBI-dependent negative growth effect in H28 cells. As shown in
Fig. 3, Cx43 AS-ODN almost
completely abolished BBI-mediated growth inhibition in H28 cells
upon down-regulation of Cx43.
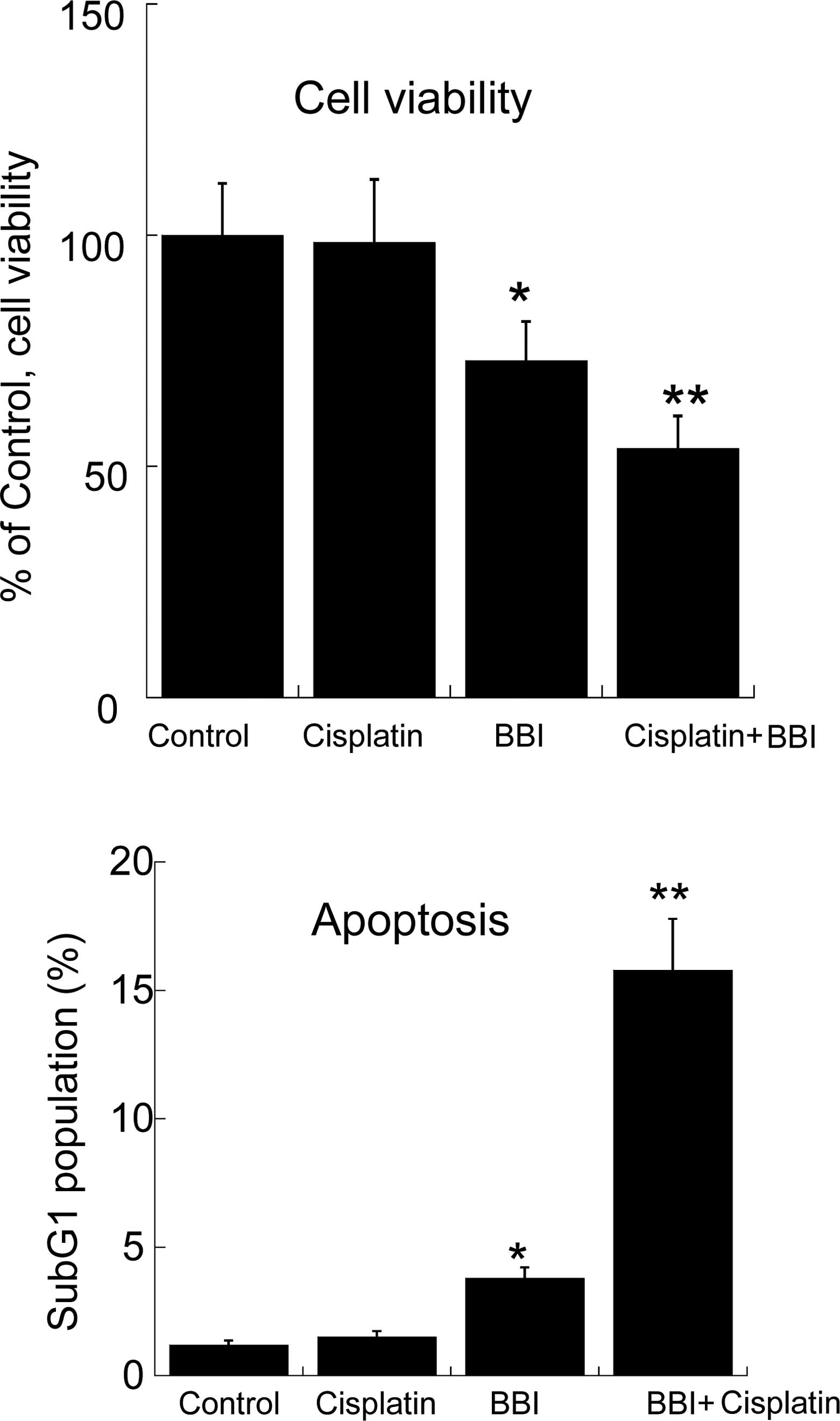
Combination effect of BBI and cisplatin
on the growth of H28 cells
We previously reported that enforced expression of
the Cx43 gene improved the resistance of cisplatin to H28 cells
(8); therefore, we investigated
whether BBI enhances cisplatin-induced cytotoxicity and apoptosis
in H28 cells. As shown in Fig. 4,
cisplatin treatment (10 μM) had no effect on cell viability in H28
cells, but the combination of BBI with cisplatin significantly
decreased the cell viability compared to BBI treatment alone. Also,
the induction of apoptosis, estimated as the subG1 population, by
the combination of BBI and cisplatin was significantly increased
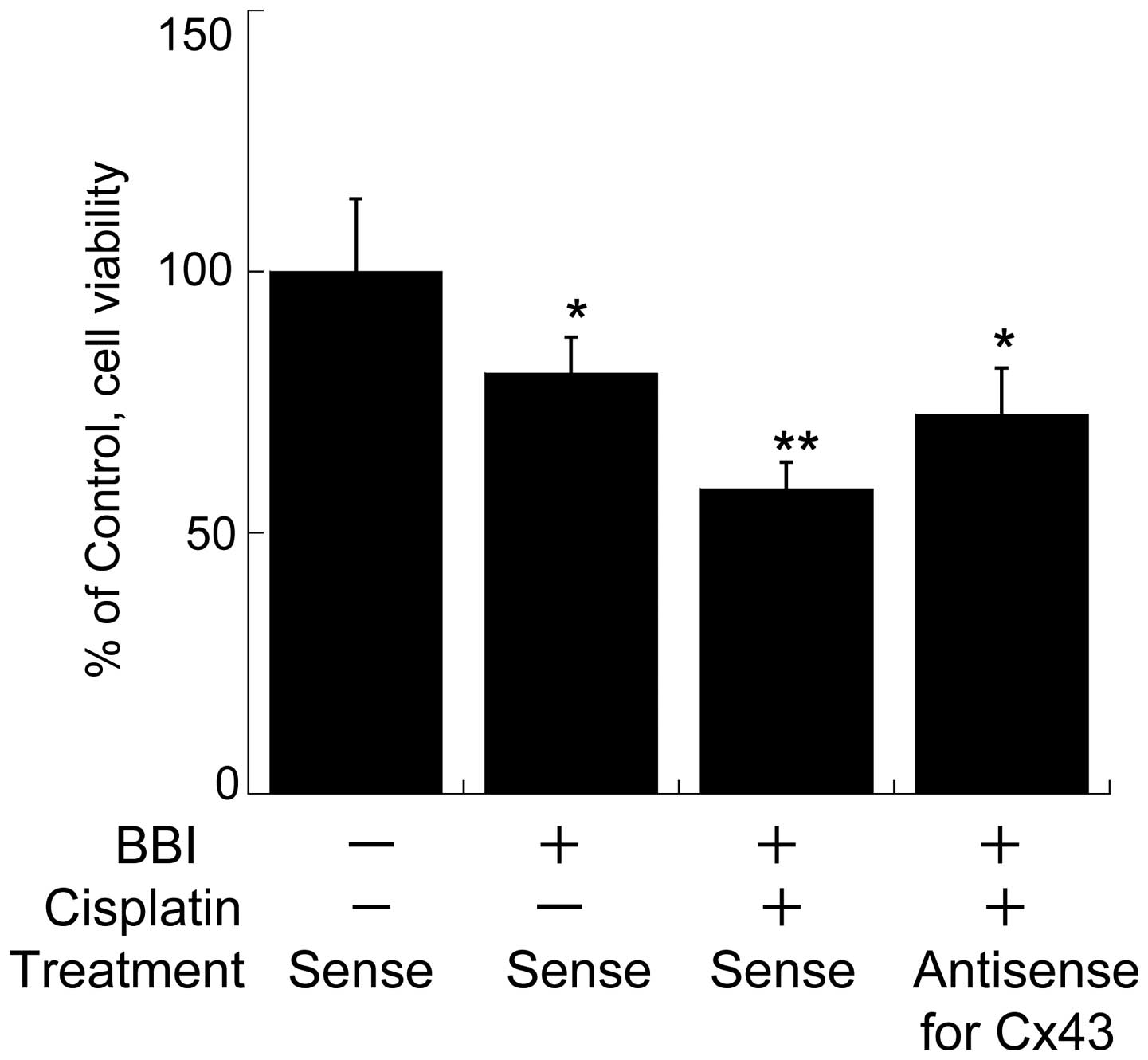
compared to that with BBI or cisplatin alone (Fig. 4). These results indicate that BBI
abrogates the resistance to cisplatin in the cells. Furthermore, in
order to ascertain whether BBI-driven restoration of Cx43 is
closely related to the improvement of chemoresistance in H28 cells
by BBI, we investigated whether AS-ODN for Cx43 cancels
BBI-enhanced cytotoxicity in H28 cells treated with cisplatin. As a
result, the enhanced effect of BBI was almost abolished by the
AS-ODN treatment (Fig. 5).
Effect of BBI on Src signaling in H28
cells
We previously reported that inactivation of the Src
family of protein tyrosine kinases is involved in enhancing the
effect of Cx43 on cisplatin-induced cytotoxicity in H28 cells
(8). Therefore, we speculated that
the enhancing effect of BBI on cytotoxicity is due to suppression
of Src signaling via the induction of Cx43. To confirm this
hypothesis, we evaluated whether BBI-enhanced Cx43 expression
improves the efficacy of cisplatin to H28 cells via the
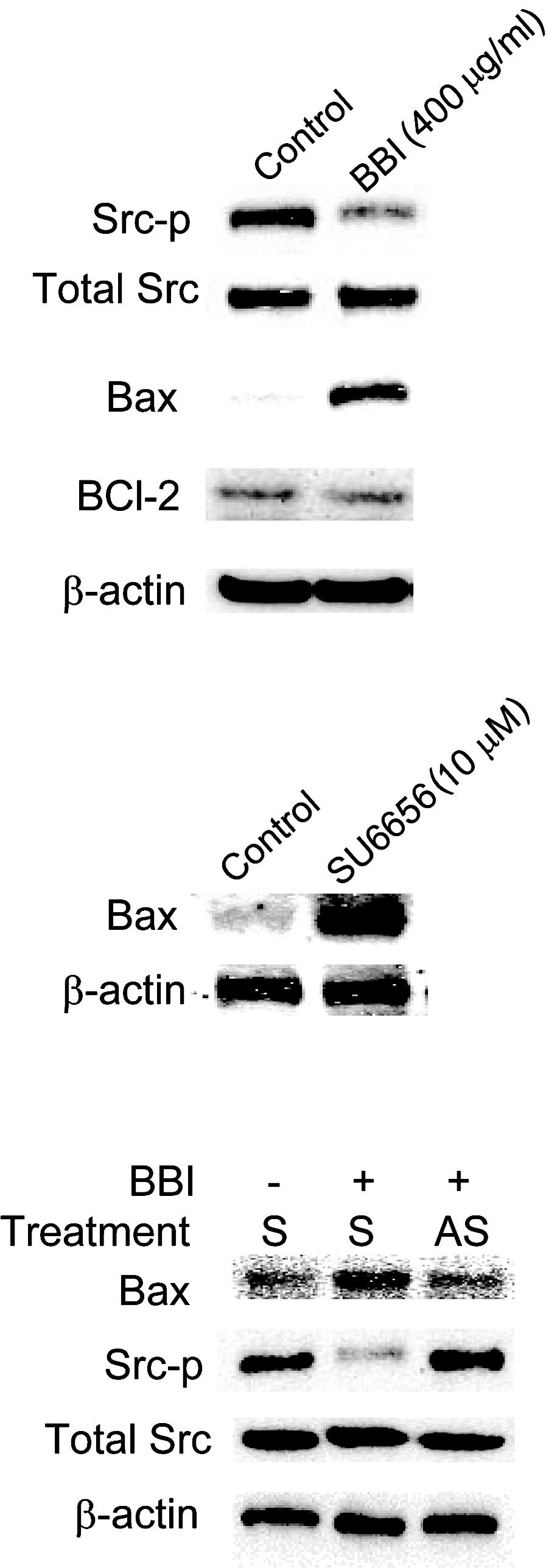
inactivation of Src signaling. As shown in Fig. 6, BBI suppressed the activation of
Src (phosphorylation on Tyr416), and knockdown of Cx43 by AS-ODN
treatment almost completely cancelled this BBI-mediated
inactivation of Src. In our previous study, Bax, a representative
pro-apoptotic factor, was shown to be induced by enforced
expression of Cx43 and to play an important role in the enhancement
of cisplatin-mediated damage in H28 cells (8); therefore, we evaluated the
contribution of Bax to the enhancing effect of BBI in H28 cells. In
the present study, BBI treatment up-regulated Bax and, upon
knockdown of Cx43 by AS-ODN treatment, the Bax level was almost
restored to the level of that in the control (Fig. 6). Additionally, the Src inhibitor,
SU6656, elevated the Bax level, indicating that inhibition of Src
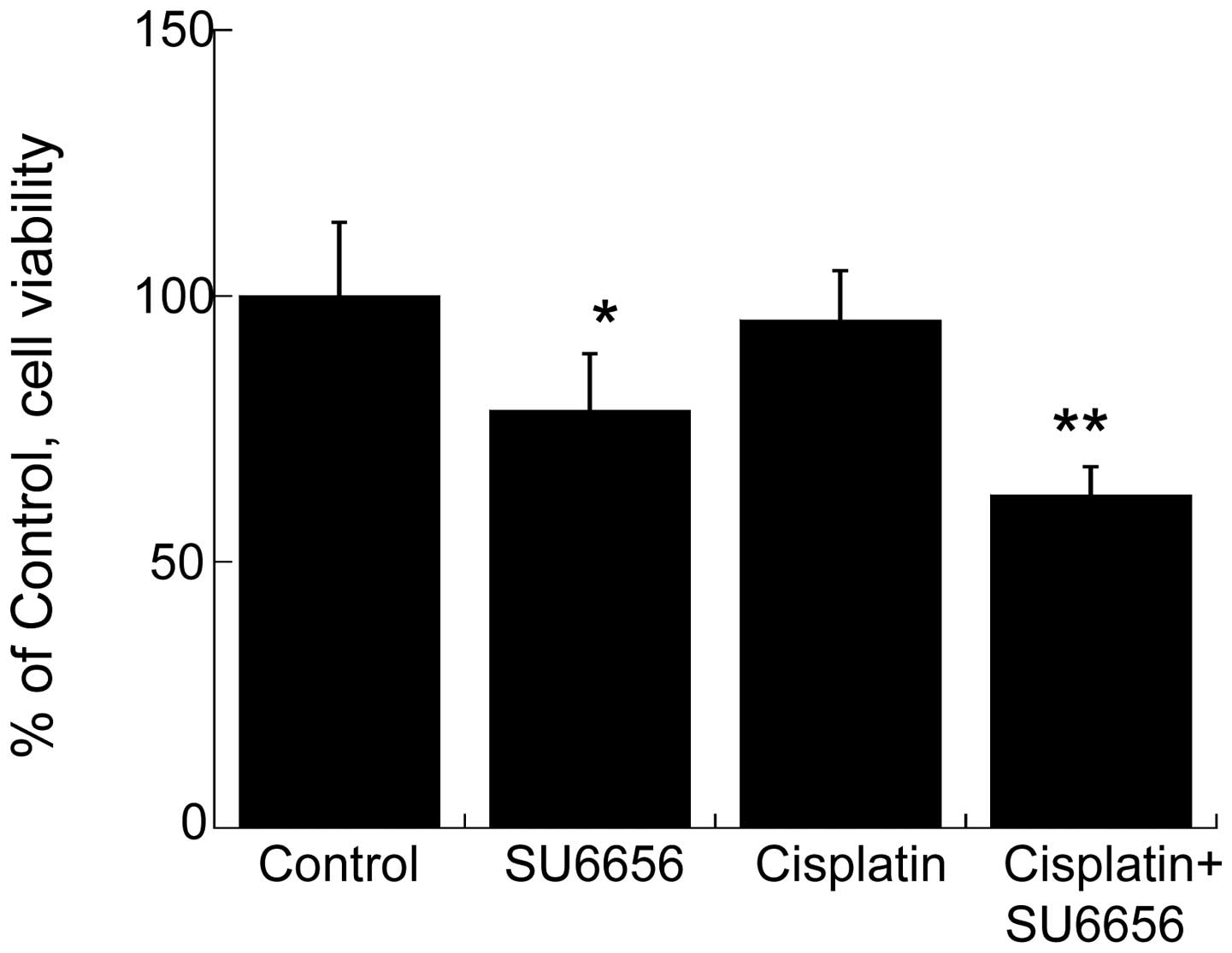
leads to the induction of Bax. Finally, in order to confirm that
Src signaling plays a crucial role in the BBI-induced enhancement
of cisplatin-mediated cytotoxicity, we evaluated the combination
effect of SU6656 and cisplatin on the growth of H28 cells. As shown
in Fig. 7, when the cells were
treated with both SU6656 and cisplatin, cell viability was
significantly inhibited as compared to that when the cells were
treated with either SU6656 or cisplatin alone.
Discussion
Management of MM continues to be a difficult
clinical issue. The relative clinical resistance to chemotherapy
and other conventional treatments strongly necessitate the
development of new potential therapeutic strategies for MM to
establish viable MM treatment protocols. In our previous study,
enforced expression of Cx43 in MM cells exhibiting resistance to
cisplatin significantly reduced this resistance (8), indicating that successful restoration
of Cx43 by a treatment agent rather than Cx43 gene transfection may
lead to the establishment of a novel effective MM therapy.
Fortunately, in regards to the restoration of Cx43 expression in
tumor cells, we and other research groups previously found that BBI
effectively caused the induction of Cx43 in several types of tumor
cells and that this induction is closely associated with negative
growth control of tumor cells (14,17).
Based on these reports, we speculated that BBI improves the
efficacy of cisplatin in MM cells via restoration of Cx43
expression. Thus, the present study was carried out to confirm this
hypothesis.
We previously reported that the Cx gene (the
molecule being Cx32) enhances the sensitivity of chemotherapeutic
agents to renal and lung cancer cells (18). Similarly, we demonstrated that
enforced Cx43 expression in H28 cells improves the effect of
cisplatin against H28 cells (14).
These reports strongly suggest that, in part, Cx-dependent tumor
suppressive effects mitigate chemoresistance in several types of
tumor cells. In the present study, we showed that BBI treatment
caused the restoration of Cx43 expression, in part, due to the
suppression of Cx43 degradation by chymotrypsin-like activity in
the proteasome, and that the restored Cx43 induced negative growth
control in H28 cells. Finally, these events caused by BBI treatment
led to an improvement in the efficacy of cisplatin. Thus, the
combination of BBI and cisplatin may be a promising new therapy
against cisplatin-resistant MM cells.
Through GJIC-dependent cell coupling, dying cancer
cells communicate cell death signals to adjacent cells which then
also die by apoptosis. These death messages which pass through the
GJ to kill cells are very likely calcium ions (19). This finding suggests that
chemoresistance to anticancer agents by tumor cells is reduced due
to the propagation of cell death signals from dying cells to
surrounding living cells via GJ. We previously reported that
inhibition of Cx-driven GJIC by a known inhibitor towards GJ
functions (18-glycyrrhetinic acid) partly abrogated
chemotherapeutic agent-induced cytotoxicity in cancer cells
(18). However, in this study we
did not find that inhibition of GJIC by the inhibitor negates the
enhanced effect of BBI on cisplatin-induced cytotoxicity in H28
cells (data not shown), indicating that BBI-mediated restoration of
GJIC does not affect cisiplatin-induced cytotoxicity. Apart from
the GJIC-dependent effect of Cx, other data showed that Cx affects
cellular homeostatic balance independently of GJIC, and that this
GJ-independent effect plays an important role in regulating
abnormal growth of cancer cells (5,20).
Moreover, we demonstrated that the Cx32 gene acts as a tumor
suppressor gene against renal cancer cells in a GJIC-independent
mechanism (21). Based on these
reports, we speculated that the GJIC-independent effect contributes
to the potentiation of cisplatin-induced cytotoxicity in H28
cells.
We previously reported that one of the
GJIC-independent tumor-suppressive effects in renal cancer cells
depends on the inactivation of Src caused by Cx32 (21). It has been well-established that
Src plays a critical role in the survival, proliferation, invasion
and metastasis of solid tumors (22), and also, the activation of Src
contributes to the appearance of malignant phenotypes in MM cells
(23). Furthermore, Src directly
and indirectly phosphorylates Cx43 at tyrosine and serine residues,
leading to loss of Cx43-mediated functions (24). On the contrary, we observed that
overexpression of Cx43 induces, not only dephosphorylation, but
also down-regulation of Src in MM cells (8). These reports suggest that these two
molecules affect their respective function based on the expression
level of each molecule. Thus, BBI-induced elevation of Cx43 may
improve the toxicity of cisplatin to H28 cells via the inactivation
of Src in a GJIC-independent manner. We confirmed that the
knockdown of Cx43 in BBI-treated H28 cells by AS-ODN abrogated the
enhancing effect of BBI on cisplatin-induced cytotoxicity, and that
Src inhibition by a specific inhibitor caused the potentiation of
the cytotoxicity. These observations completely support the above
speculation.
In general, the existence of anti-apoptotic
molecules and pro-apoptotic molecules is well known (25). Bcl-2 is a major anti-apoptotic
protein which protects cells from a wide variety of apoptotic
stimuli (26). By contrast, Bax is
a Bcl-2-like protein that binds to and antagonizes the protective
effect of Bcl-2, rendering cells more sensitive to apoptosis
(27). In the present study, we
found that only the level of Bax in H28 cells was significantly
elevated by BBI treatment. As a result, the balance between pro-
and anti-apoptotic factors was altered towards the induction of
apoptosis. Any approach that alters the balance in favor of
apoptosis may thus confer a therapeutic benefit. Overall, our
present results suggest that Cx43 induced by BBI treatment
influences the balance between pro- and anti-apoptotic factors in
the direction of apoptosis, possibly contributing to the improved
sensitivity of H28 cells to cisplatin. At present, the reason why
BBI enhanced the level of Bax remains unclear. From the present
data, we can only speculate that the inactivation of Src signaling
caused by BBI-induced elevation of Cx43 was associated with the
up-regulation of Bax. Since it is known that Bax is located
downstream in the Src signal pathway, it appears that the
inhibition of Src signaling indirectly induces Bax in H28 cells
treated with BBI. In order to clarify the mechanism by which the
enhancing effect of BBI on cisplatin-induced cytotoxicity in H28
cells is activated, further study is warranted.
Acknowledgements
This study was supported by a research
grant for Health Sciences Focusing on Drug Innovation from the
Japan Health Sciences Foundation (KHC1023), and a grant-in-aid for
Science Research from the Ministry of Education, Culture and
Sciences of Japan (no. 22500754) to Yano T.
References
|
1.
|
Carbone M, Kratzke RA and Testa JR: The
pathogenesis of mesothelioma. Semin Oncol. 29:2–17. 2002.
View Article : Google Scholar
|
|
2.
|
Nowak AK, Lake RA, Kindler HL and Robinson
BW: New approaches for mesothelioma: biologics, vaccines, gene
therapy, and other novel agents. Semin Oncol. 29:82–96. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ryan CW, Herndon J and Vogelzang NJ: A
review of chemotherapy trials for malignant mesothelioma. Chest.
113:S66–S73. 1998. View Article : Google Scholar
|
|
4.
|
Van Haarst JM, Baas P, Manegold CH, et al:
Multicentre phase II study of gemcitabine and cisplatin in
malignant pleural mesothelioma. Br J Cancer. 86:342–345.
2002.PubMed/NCBI
|
|
5.
|
Vinken M, Vanhaecke T, Papeleu P, et al:
Connexins and their channels in cell growth and cell death. Cell
Signal. 18:592–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Laird DW: Life cycle of connexins in
health and disease. Biochem J. 394:527–543. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mesni M, Krutovskikh V, Piccoli C, et al:
Negative growth control of HeLa cells by connexin genes: connexin
species specificity. Cancer Res. 55:629–639. 1995.PubMed/NCBI
|
|
8.
|
Sato H, Iwata H, Takano Y, Yamada R,
Okuzawa H, Nagashima Y, Yamaura K, Ueno K and Yano T: Enhanced
effect of connexin 43 on cisplatin-induced cytotoxicity in
mesothelioma cells. J Pharm Sci. 110:466–475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kennedy AR: The Bowman-Birk inhibitor from
soybeans as an anticarcinogenic agent. Am J Clin Nutri.
68:S1406–S1412. 1998.PubMed/NCBI
|
|
10.
|
Armstrong WB, Kennedy AR, Wan XS, et al:
Single-dose administration of Bowman-Birk inhibitor concentrate in
patients with oral leukoplakia. Cancer Epid Biomark Prev. 9:43–47.
2000.PubMed/NCBI
|
|
11.
|
Billings PC, Newberne PM and Kennedy AR:
Protease inhibitor suppression of colon and anal gland
carcinogenesis induced by dimethylhydrazine. Carcinogenesis.
11:1083–1086. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kennedy AR: Prevention of carcinogenesis
by protease inhibitors. Cancer Res. 54:S1999–S2005. 1994.PubMed/NCBI
|
|
13.
|
Suzuki K, Yano T, Sadzuka Y, Sugiyama T,
Seki T and Asano R: Restoration of connexin 43 by Bowman-Birk
protease inhibitor in M5076-bearing mice. Oncol Rep. 13:1247–1250.
2005.PubMed/NCBI
|
|
14.
|
Saito T, Sato H, Virgona N, Hagiwara H,
Kashiwagi K, Suzuki K, Asano R and Yano T: Negative growth control
of osteosarcoma cells by Bowman-Birk protease inhibitor from
soybean; involvement of connexin 43. Cancer Lett. 253:249–257.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Nishimura M, Saito T, Yamasaki H and Kudo
R: Suppression of gap junctional intercellular communication via 5′
CpG island methylation in promoter region of E-cadherin gene in
endometrial cancer cells. Carcinogenesis. 24:1615–1623. 2003.
|
|
16.
|
Nam S, Smith DM and Dou QP: Ester
bond-containing tea polyphenols potently inhibit proteasome
activity in vitro and in vivo. J Biol Chem. 276:13322–13330. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Tang M, Asamoto M, Ogawa K, et al:
Induction of apoptosis in the LNCaP human prostate carcinoma cell
line and prostate adenocarcinomas of SV40T antigen transgenic rats
by the Bowman-Birk inhibitor. Pathol Int. 59:790–796. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sato H, Senba H, Virgona N, Fukumoto K,
Ishida T, Hagiwara H, Negishi E, Ueno K, Yamasaki H and Yano T:
Connexin 32 potentiates vinblastine-induced cytotoxicity in renal
cell carcinoma cells. Mol Carcinog. 46:215–224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Krutovskikh VA, Piccoli C and Yamasaki H:
Gap junction intercellular communication propagates cell death in
cancerous cells. Oncogene. 21:1989–1999. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Duflot-Dancer A, Mesnil M and Yamasaki H:
Dominant-negative abrogation of connexin-mediated cell growth
control by mutant connexin genes. Oncogene. 15:2151–2158. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fujimoto E, Sato H, Shirai S, Nagashima Y,
Fukumoto K, Hagiwara H, Negishi E, Ueno K, Omori Y, Yamasaki H,
Hagiwara K and Yano T: Connexin32 as a tumor-suppressor gene in a
metastatic renal cell carcinoma cell line. Oncogene. 24:3684–3690.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Benati D and Baldari CT: SRC family
kinases as potential therapeutic targets for malignancies and
immunological disorders. Curr Med Chem. 15:1154–1165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Tsao AS, He D, Saigal B, et al: Inhibition
of c-Src expression and activation in malignant pleural
mesothelioma tissues leads to apoptosis, cell cycle arrest, and
decreased migration and invasion. Mol Cancer Ther. 6:1962–1972.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Pahujaa M, Anikin M and Goldberg GS:
Phosphorylation of connexin43 induced by Src: regulation of gap
junctional communication between transformed cells. Exp Cell Res.
313:4083–4090. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kim R, Emi M, Tanabe K, et al: Therapeutic
potential of antisense Bcl-2 as a chemosensitizer for cancer
therapy. Cancer. 101:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Chao DT and Korsmeyer SJ: BCL-2 family:
regulators of cell death. Ann Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar
|
|
27.
|
Hockenbery DM: Targeting mitochondria for
cancer therapy. Environ Mol Mutagenesis. 51:476–489. 2010.
View Article : Google Scholar : PubMed/NCBI
|