Introduction
The pathogenesis of liver cirrhosis is complex,
involving several signal transduction pathways and several liver
cell types (1). Stellate cells,
non-parecnhymal cells in the liver, become activated during
liver injury and initiate programs of transforming growth factor
(TGF)-β1 production and extracellular matrix (ECM) remodeling that
contribute to the apoptosis of hepatocytes (2). Ethanol injury leads to apoptosis of
hepatocytes and an increase in the production of fibrogenic
cytokines, including TGF-β1. TGF-β1 has also been revealed to
induce de-differentiation and epithelial-mesenchymal transition
(EMT) in hepatocytes (3).
The bone morphogenetic proteins (BMPs) are a large
class of multifunctional growth factors and are involved in a major
developmental signaling pathway critical for embryogenesis and
tissue generation in organs, including the kidney and lung
(4). However, BMPs are also
essential during postnatal life and regulate cell proliferation,
differentiation, apoptosis, angiogenesis and the secretion of ECM
components (5). BMP-7 is
considered to have inhibitory effects since it is able to
counteract TGF-β1-induced fibrotic effects in vitro and
reverse established fibrosis in organs as diverse as the kidney,
heart and colon (6).
Alendronate sodium is a bisphosphonate that acts as
a specific inhibitor of osteoclast-mediated bone resorption.
Bisphosphonates are synthetic analogs of pyrophosphate which bind
to hydroxyapatite in bone. Alendronate sodium is chemically
described as (4-amino-1-hydroxybutylidene) bisphosphonic acid
monosodium salt trihydrate. Alendronate sodium is a white,
crystalline, non-hygroscopic powder (7).
The present study demonstrates for the first time
that alendronate sodium significantly arrests the progression of
hepatic fibrosis. The underlying mechanism was associated with
changes in the redox state and involved marked decreases in the
expression of TGF-β1 and α-smooth muscle actin (α-SMA) and
upregulation of the expression of BMP-7 and E-cadherin in liver
tissue.
Materials and methods
Chemicals and materials
Glass slides (75×25 mm2) were obtained
from Gibco (Carlsbad, CA, USA) and (3-acryloxypropyl)
trichlorosilane was purchased from Gelest, Inc. (Morrisville, PA,
USA). Streptavidin-conjugated Alexa 546, AlexaFluor 488 anti-mouse
IgG, BMP-7 and TGF-β1 were obtained from Sigma-Aldrich (St. Louis,
MO, USA). Mouse anti-E-cadherin antibody was purchased from BD
Biosciences (Franklin Lakes, NJ, USA). Concentrated
phosphate-buffered saline (10X PBS) was purchased from Lonza
(Shanghai, China). Minimal essential medium (MEM), sodium pyruvate,
nonessential amino acids, fetal bovine serum (FBS), Superscript
III, RNaseOut (RNase inhibitor) and dNTPs were purchased from
Invitrogen (Shanghai, China). Polypropylene microarray plates (384
well) were obtained from Genetix (Shanghai, China). Goat anti-mouse
cross-adsorbed albumin antibody was obtained from Sigma-Aldrich.
Formalin was purchased from Fisher (Shanghai, China). The ApopTag
Red in situ apoptosis detection kit was obtained from
Chemicon (Shanghai, China). DAPI stain mounting media were
purchased from Vectorshield (Shanghai, China).
Animals
Adult gender-matched (n=20 each gender) C57BL mice
weighing 200±10.2 g were purchased from Tongji University
Laboratories (Shanghai, China) and fed on a commercial diet with
water. All animal experiments were performed according to the
National Institute of Health (NIH) guidelines for the ethical care
and use of laboratory animals and the study was approved by the
Tongji Animal Care and Use Committee of China.
Drugs
The alendronate sodium tablets (10 mg) also
contained carnauba wax. Each bottle of the oral solution contained
91.35 mg alendronate monosodium salt trihydrate, which was the
molar equivalent of 70 mg free acid.
Groups
Adult numbered mice (n=40) were assigned randomly to
one of four groups. In the normal control group, 10 mice received
intraperitoneal injections of olive oil (0.5 ml/100 mg) twice each
week. In the alendronate sodium control group, 10 mice received
intraperitoneal injections of olive oil (0.5 ml/100 mg) and
alendronate sodium (25 mg/kg) at the same time, twice each week. In
the liver fibrosis model group, 10 liver fibrosis model mice
received intraperitoneal injections of 40% CCl4 and
olive oil mixture (0.5 ml/100 mg, Sigma-Aldrich) as previously
described (8). In the alendronate
sodium-treated group, 10 mice received intraperitoneal injections
of 40% CCl4 and olive oil admixture (0.5 ml/100 mg)
twice each week, as well as alendronate sodium (25 mg/kg) at the
same time. The mice were sacrificed after 8 weeks of treatment.
Cell isolation and culture
Liver epithelial cells were isolated from normal
Sprague-Dawley mice as follows: following in situ perfusion
of the liver with pronase (Roche, Indianapolis, IN, USA) and
collagenase (Roche), dispersed cell suspensions were layered in a
discontinuous density gradient of 5.8% Larcoll (Sigma-Aldrich) and
15.6% Histodenz (Sigma-Aldrich). The resulting upper layer
consisted of >98% liver epithelial cells. The purity and
viability were verified by phase-contrast microscopy with
examination of autofluorescence and propidium iodide exclusion (50
μg/ml; Roche). Liver epithelial cells were cultured in 10%
serum-supplemented DMEM (Invitrogen) with
streptomycin-penicillin.
Reverse transcription and real-time
quantitative PCR (RT-PCR) analysis
PBMC solution (100 μl) priority was added to
300 μl Trizol lysate and 100 μl chloroform at 4°C,
and spun at 1,3200 rpm for 15 min. The liquid supernatant
affiliated isopyknic avantin, −20°C deposit 30 min, 4°C 1,3200 rpm
for 15 min. The liquid supernatant was added to 300 μl 75%
alcohol at 4°C and spun at 1,3200 rpm for 15 min. The liquid
supernatant was added to diethyl carbonate (DEPC). This solution
was used as the total RNA solution of PBMC. Total RNA solution (10
μl) with 1 μl random primers (0.2
μg/μl) and 1 μl DEPC was denatured at 70°C for
5 min, then mixed with 4 μl 5X RT buffer, 1 μl RNasin
(20 μg/μl), 2 μl 10 mM dNTP mix and 1
μl reverse transcription enzyme M-MLV (20
μg/μl) at 42°C for 60 min. Next, the solution was
heated to 70°C for 10 min, then cooled in ice water and stored at
−20°C. The RT-PCR used a 20-μl reaction system with 2
μl RNase inhibitor, 7.2 μl deionized water, 10
μmol/l forward and reverse primers (0.4 μl each) and
10 μl 2X RT-PCR MasterMix (bulk volume, 20 μl). In
the RT-PCR Master Mix SYBR-Green I dye fluorescence signal
intensity was associated with the quantity of DNA and 55 point
fluorescence was set-up as the monitoring point. The comparative Ct
value method, using a housekeeping gene (GAPDH) as an internal
standard, was employed to determine the relative levels of TGF-β1,
α-SMA, N-cadherin, fibroblast-specific protein 1 (FSP1, also called
S100A4), BMP-7 and E-cadherin.
Western blotting
Whole cell proteins (20 μg) were separated by
PAGE and transferred to nylon membranes. The primary antibodies
were as follows: anti-α-SMA (1:2,000 dilution; Dako, Carpinteria,
CA, USA), anti-TGF-β1 (1:2,000 dilution; Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA), anti-E-cadherin (1:2,000 dilution;
USA), anti-BMP-7 (1:1,500 dilution; Cell Signaling Technology,
Danvers, MA, USA), anti-FSP1 (1:2,000 dilution; Santa Cruz
Biotechnology) and anti-GAPDH (1:2,000 dilution; Sigma-Aldrich).
Appropriate secondary antibodies were used and antigens were
detected by enhanced chemiluminescence (Pierce Biotechnology,
Rockford, IL, USA).
Statistical analyses
The SPSS 12.0 software (SPSS, Chicago, IL, USA) was
used for statistical analysis. Quantitative variables of normality
were tested and if the data conformed to a normal distribution they
were expressed as means ± SD. Two independent t-tests were
performed and if the data were not normally distributed, they were
expressed as medians with a range and non-parametric tests were
considered. For categorical data, non-parametric tests were used to
compare frequencies. P<0.001 (two-sided) was considered to
indicate statistically significant differences.
Results
Aspartate aminotransferase (AST) and
alanine aminotransferase (ALT) detection
ALT and AST activities were significantly increased
in the liver fibrosis model group compared with those in the normal
control (P<0.001). In the alendronate sodium-treated group, the
ALT and AST activities were markedly reduced compared with those in
the liver fibrosis mice which were not treated with alendronate
sodium (P<0.001).
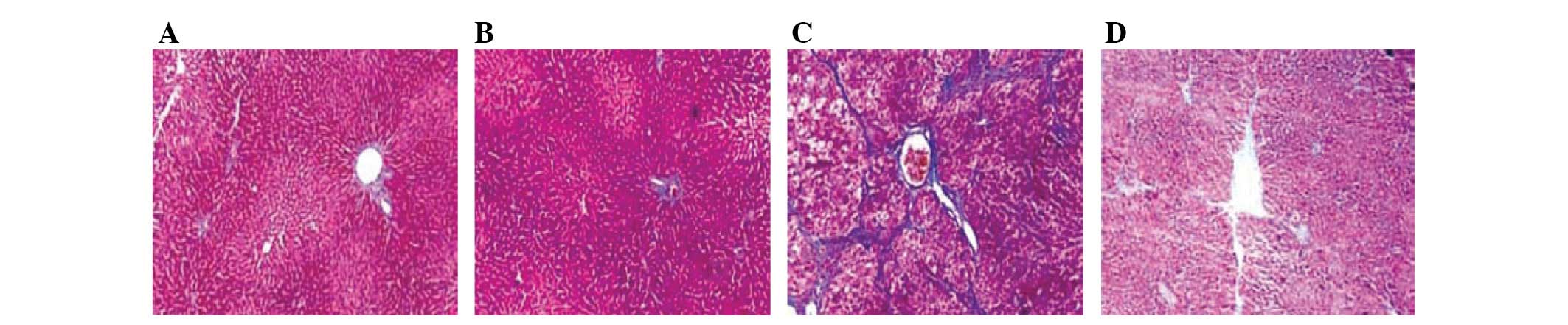
Alendronate sodium-treated pathology
expressed in mouse liver fibrosis
Following alendronate sodium (25 mg/kg) treatment
twice each week for 8 weeks, hepatic tissue sections stained with
Masson’s trichrome stain exhibited a reversal of mouse liver
fibrosis to a significant extent (Fig.
1).
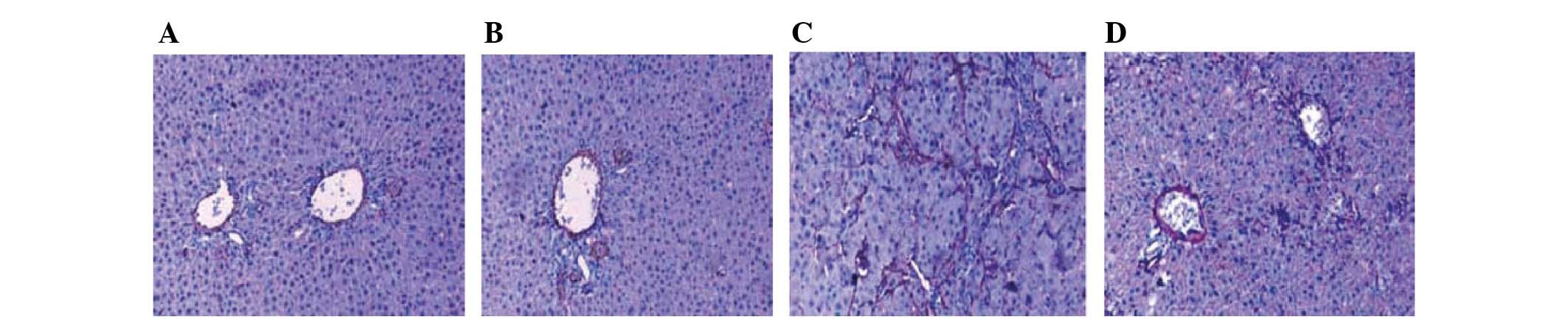
Expression of α-SMA
Following alendronate sodium treatment, the
expression of α-SMA positive cells in the hepatic fibrosis area was
significantly reduced (P<0.05; Fig.
2).
Expression of TGF-β1, α-SMA, N-cadherin,
FSP1, BMP-7 and E-cadherin mRNA
In the mouse liver fibrosis model, increased
expression of TGF-β1, α-SMA, N-cadherin and FSP1 mRNA was observed,
but the expression of of BMP-7 and E-cadherin was reduced. The
opposite expression pattern was observed in the alendronate
sodium-treated group (Fig. 3).
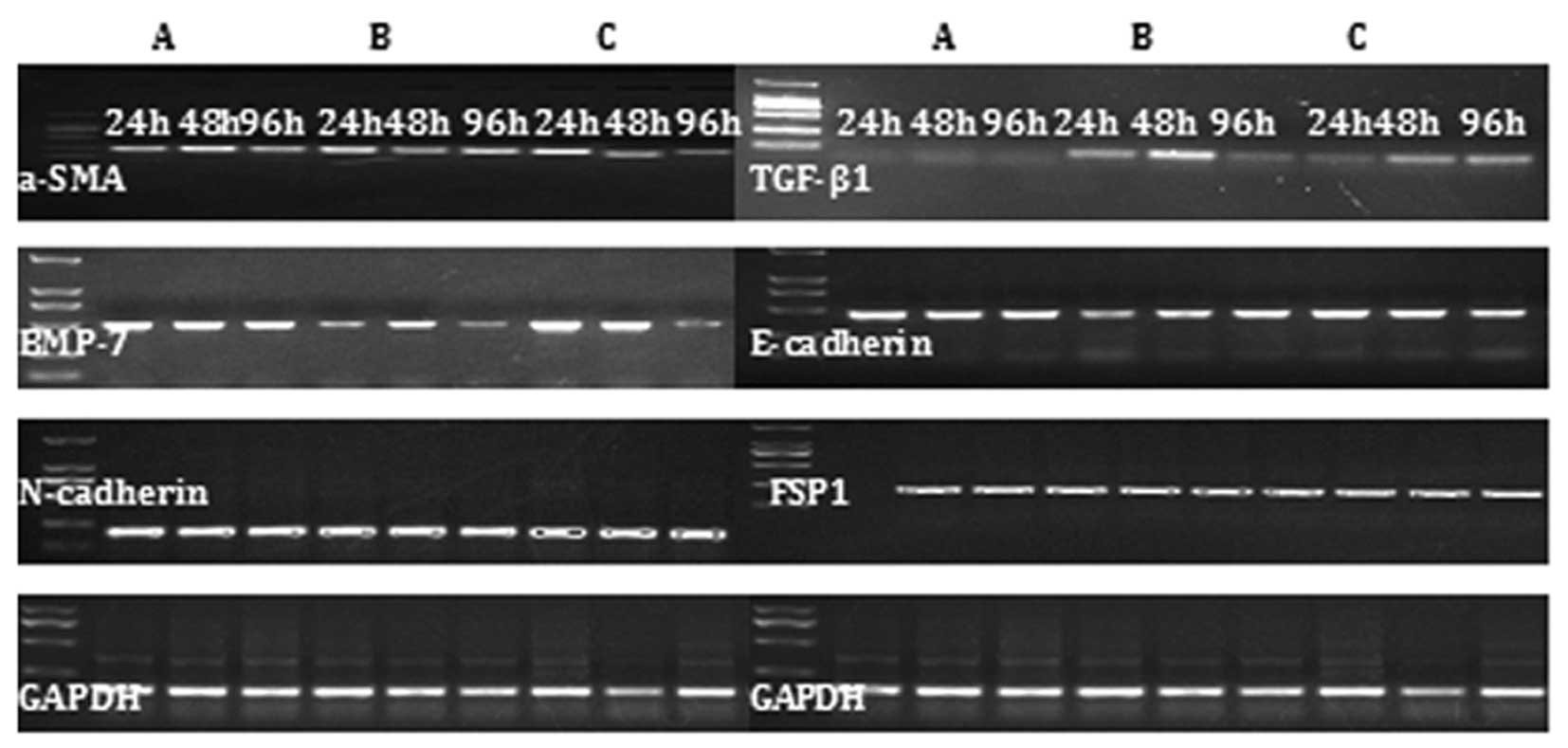 | Figure 3RT-PCR results demonstrating the
effect of CCl4-induced mouse liver fibrosis on the
expression of TGF-β1, α-SMA, N-cadherin, FSP1, BMP-7 and E-cadherin
mRNA during EMT in (A) the control group, (B) the liver fibrosis
model group and (C) the alendronate sodium-treated group during
chronic exposure to TGF-β1 (5 ng/ml) from 24 to 96 h. TGF-β1,
transforming growth factor-β1; α-SMA, α-smooth muscle actin; FSP1,
fibroblast-specific protein 1; BMP-7, bone morphogenetic protein-7;
EMT, epithelial-mesenchymal transition. |
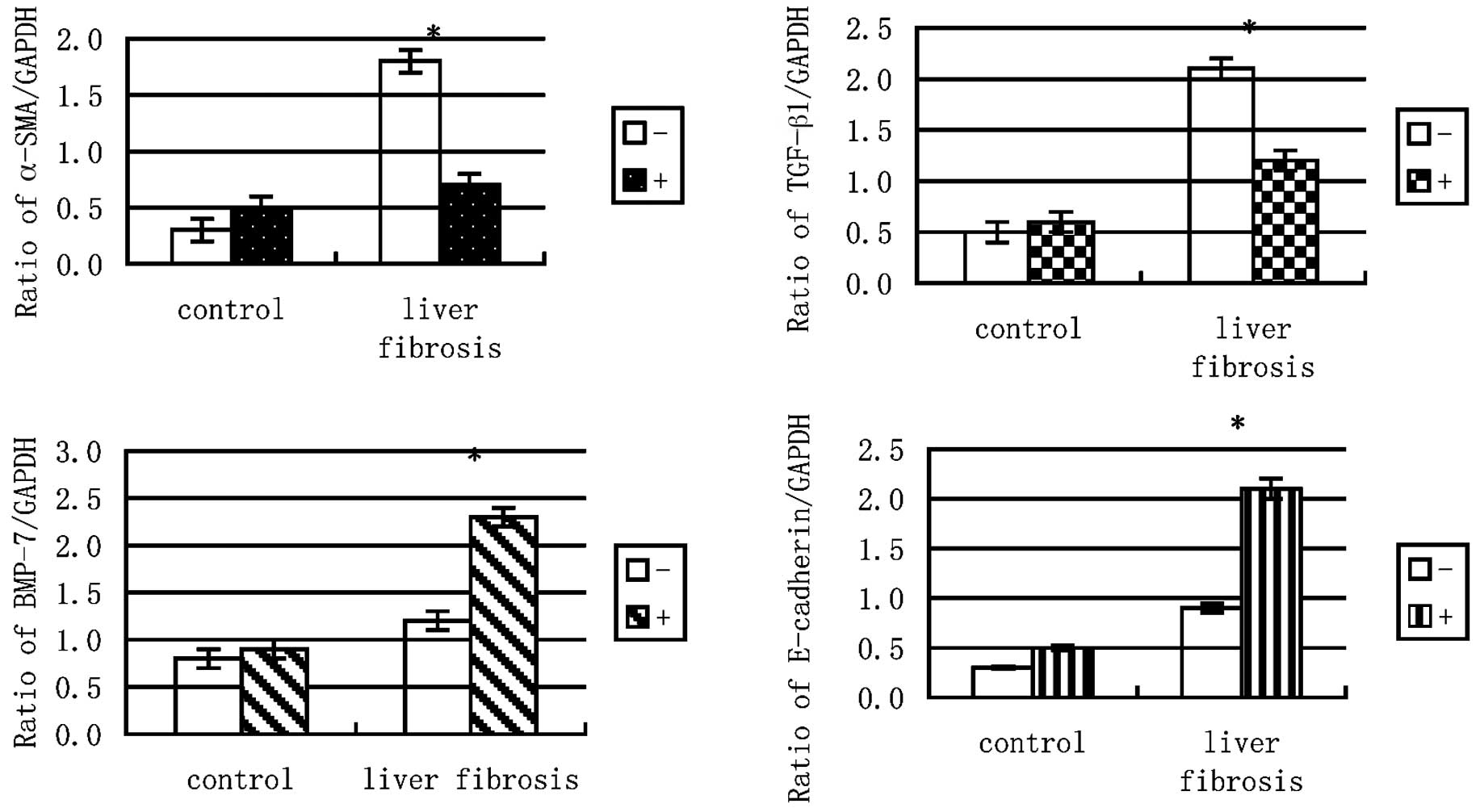
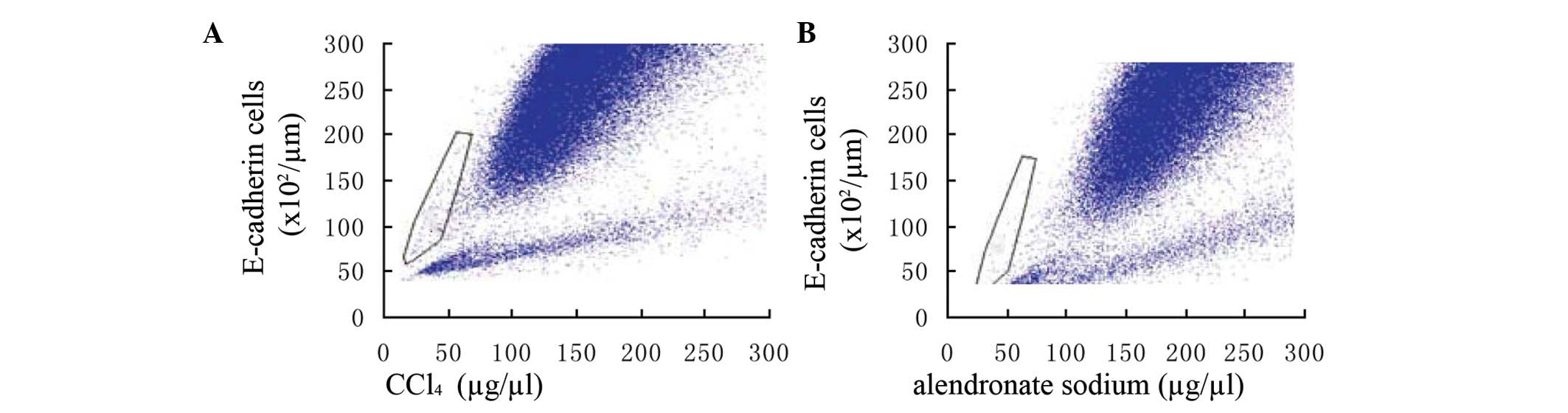
Expression ratios of TGF-β1, α-SMA, BMP-7
and E-cadherin to GAPDH mRNA
Chronic exposure to TGF-β1 (5 ng/ml) for 96 h
increased the expression ratios of α-SMA and TGF-β1 to GAPDH but
following alendronate sodium treatment, the expression ratio of
α-SMA to GAPDH decreased. The BMP-7 and E-cadherin to GAPDH ratios,
however, were increased further by the alendronate sodium treatment
(Fig. 4).
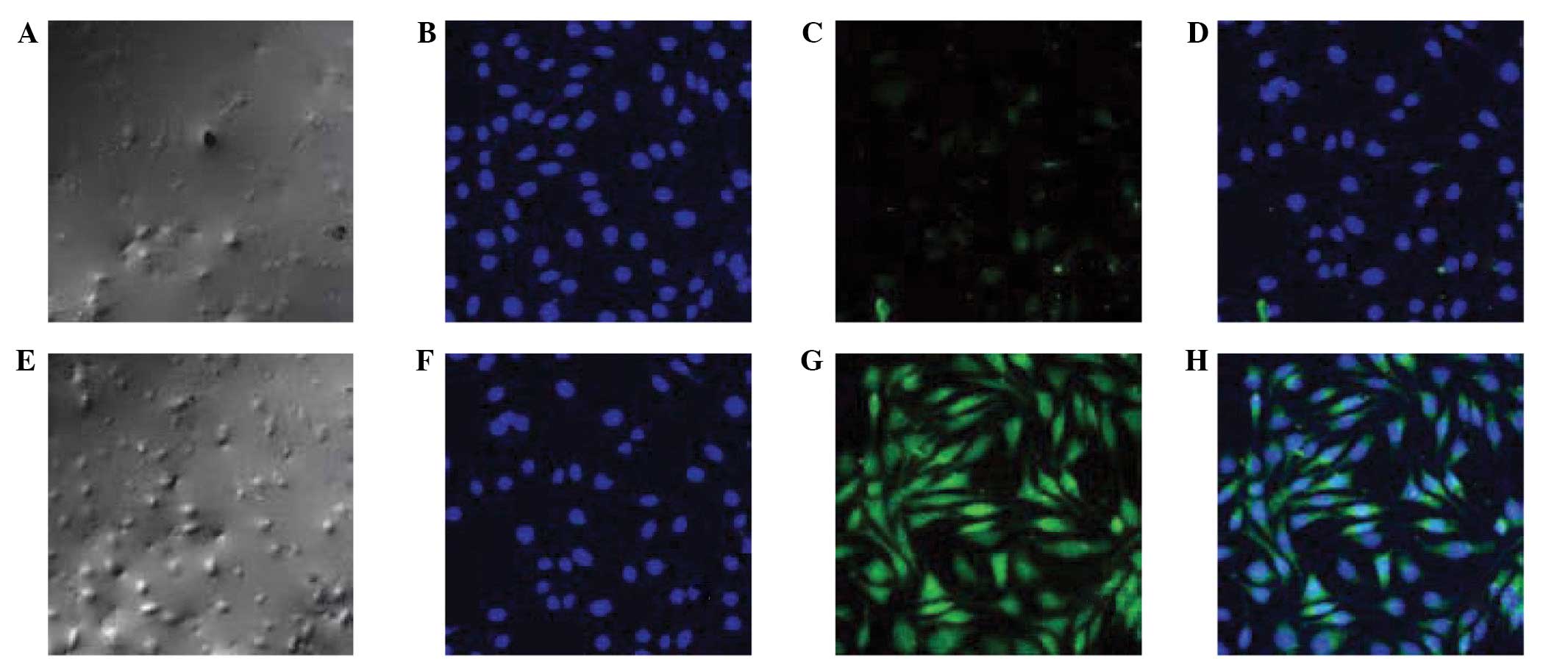
Protein expression of E-cadherin and
α-SMA in each group
Following alendronate sodium treatment, the protein
expression of E-cadherin (green fluorescence) was denser, while
that of α-SMA (blue fluorescence) was more scattered than in the
controls (Fig. 5).
Analysis of E-cadherin in alendronate
sodium-treated cells
Following alendronate sodium treatment, the
expression of E-cadherin in the cells of the hepatic fibrosis area
was greater and denser than in the controls. (Fig. 6)
Discussion
The effects of TGF-β1-induced EMT on the structure,
migration, cytoskeletal dynamics and long-term correlations of
mammalian epithelial cell lines have been investigated with
time-resolved impedance analysis (8). Liver sections were labeled to detect
antigens associated with biliary epithelial cells (E-cadherin), EMT
[FSP1, vimentin, N-cadherin, matrix metalloproteinase (MMP)-2 and
α-SMA] and intracellular signal-transduction mediated by
phosphorylated (p) Smad 2/3 (9).
The mechanisms underlying the morphological and
phenotypic changes of epithelial markers undergoing EMT in liver
fibrosis include a loss of E-cadherin and cytokeratin; increased
expression of FSP1, vimentin, N-cadherin and α-SMA; basement
membrane component loss; and the production of interstitial-type
matrix molecules, including fibronectin and type I/III collagen
(10).
In the present study, significantly less α-SMA and
TGF-β1 and more BMP-7 and E-cadherin was expressed in the
alendronate sodium-treated group and the controls than in the
CCl4-induced mouse liver fibrosis model group
(P<0.001). Following the exposure of these primary mouse
hepatocytes to 5 ng/ml TGF-β1 for 96 h, the alendronate
sodium-treated group exhibited increased staining for the
epithelial markers E-cadherin which was accompanied by the
decreased expression of various mesenchymal markers, including
α-SMA and FSP-1. Alendronate sodium may also affect the activities
of several signaling pathways that trigger the EMT, such as the
Notch, Wnt and integrin pathways (11). The present study demonstrated that
in the liver fibrosis model mice the expression of TGF-β1 and α-SMA
was increased. However, in the alendronate sodium-treated group
this was accompanied by the decreased expression of TGF-β1 and
α-SMA, but increased the expression of BMP-7 and E-cadherin
observed by western blotting.
The administration of alendronate, a potent
inhibitor of bone resorption, was observed to be associated with an
increase in the bone mineral density. Alendronate also reduced the
activity of the parathyroid hormone which also stimulates bone
resorption, thereby releasing preformed growth factors that are
adsorbed to the bone matrix, such as insulin-like growth factor 1
and TGF-β1 (12). However, the
role of alendronate in liver fibrosis remains unclear. Alendronate
may regulate tissue remodeling by controlling TGF-β1-induced
profibrotic functions in liver fibrosis. Thus the data of the
present study suggest that EMT occurred in mouse liver fibrosis and
induced the accumulation of TGF-β1-and α-SMA-expressing mesodermal
cells while expanding the endodermal compartment during liver
morphogenesis, suggesting that alendronate may also aid the
reversal of mouse liver fibrosis (13).
EMT is induced by the integrated actions of numerous
stimuli, including TGF-β1 and matrix-generated signals that are
also known to be implicated in inflammation, repair responses and
fibrosis (14). Chronic exposure
to TGF-β1 induces the transition of hepatocytes to
collagen-producing mesenchymal cells and the prolonged exposure of
hepatocytes to TGF-β1 increases the expression of collagen and
induces cytoskeletal rearrangement that resembles the EMT (15). These morphological and molecular
alterations may provide the foundation for liver fibrosis.
Consideration of the association and mechanisms of
EMT and alendronate in liver fibrosis suggests that the underlying
mechanism is associated with changes in the redox state and that
alendronate markedly decreased the expression of TGF-β1 and α-SMA
in the liver tissue. The redox state may remain variable in liver
fibrosis and depends on the balance between TGF-β/smad- and
BMP-7-modulated mechanisms that regulate EMT and
mesenchymal-epithelial transition (MET) in multifunctional
progenitors. In all these processes, TGF-β1 acts profibrogenically
while alendronate has opposing effects. The balance of these
cytokines is further modulated by TGF-β1 which reduces alendronate
activities. This may explain the mechanism of hepatic fibrosis. EMT
may be important for the diagnosis of hepatic fibrosis and for
developing studies of the pathogenesis of hepatic fibrosis and
establishing effective preventive approaches.
The present study revealed that alendronate reduces
the ability of TGF-β1 to increase the induction of EMT in liver
fibrosis which was consistent with the hypothesis that TGF-β1
signaling induces the EMT through various signaling mechanisms and
is the predominant agent mediating these fibrotic changes.
Alendronate was identified following previous descriptions
characterising its biological activity in extracts of demineralised
bone. Alendronate also synthesises a number of growth regulatory
peptides which are stored in the bone matrix and are possibly
responsible for normal bone formation.
Abbreviations:
|
TGF-β1
|
transforming growth factor-β1;
|
|
BMP-7
|
bone morphogenetic protein-7;
|
|
EMT
|
epithelial-mesenchymal transition;
|
|
MET
|
mesenchymal-epithelial transition;
|
|
α-SMA
|
α-smooth muscle actin
|
Acknowledgements
This study was supported by a grant
(No. 81070343) from the National Natural Science Foundation of
China and partly by a grant from the Shanghai Excellent Academic
Leaders Program (No. 08D14045).
References
|
1
|
Lee JG and Kay EP: NFκB is the
transcription factor for FGF-2 that causes endothelial mesenchymal
transformation in cornea. Invest Ophthalmol Vis Sci. 53:1530–1538.
2012.
|
|
2
|
Bi WR, Yang CQ and Shi Q: Transforming
growth factor-β1 induced epithelial-mesenchymal transition in
hepatic fibrosis. Hepato-Gastroenterology. 59:191–194. 2012.
|
|
3
|
Zhang Y, Wei J, Wang H, et al: Epithelial
mesenchymal transition correlates with CD24+CD44+ and CD133+ cells
in pancreatic cancer. Oncol Rep. 27:1599–1605. 2012.
|
|
4
|
Huse K, Bakkebø M, Wälchli S, et al: Role
of Smad proteins in resistance to BMP-induced growth inhibition in
B-cell lymphoma. PLoS One. 7:e461172012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bi WR, Yang CQ and Jin CZ: Epithelial
mesenchymal transition play an important role in hepatic fibrosis.
J Tongji Univ. 33:113–116. 2012.(In Chinese).
|
|
6
|
Mathias RA, Ji H and Simpson RJ: Proteomic
profiling of the epithelial mesenchymal transition using 2D DIGE.
Methods Mol Biol. 854:269–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luo W, Fang W, Li S and Yao K: Aberrant
expression of nuclear vimentin and related epithelial-mesenchymal
transition markers in nasopharyngeal carcinoma. Int J Cancer.
131:1863–1873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
García de Herreros A and Baulida J:
Cooperation, amplification, and feed-back in epithelial-mesenchymal
transition. Biochim Biophys Acta. 1825:223–228. 2012.PubMed/NCBI
|
|
9
|
Lim M, Chuong CM and Roy-Burman P: PI3K,
Erk signaling in BMP7-induced epithelial-mesenchymal transition
(EMT) of PC-3 prostate cancer cells in 2- and 3-dimensional
cultures. Horm Cancer. 2:298–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizuiri S, Hemmi H, Arita M, et al:
Effluent markers related to epithelial mesenchymal transition with
adjusted values for effluent cancer antigen 125 in peritoneal
dialysis patients. Int J Nephrol. 2011:2610402011. View Article : Google Scholar
|
|
11
|
Bramlage CP, Tampe B, Koziolek M, et al:
Bone morphogenetic protein (BMP)-7 expression is decreased in human
hypertensive nephrosclerosis. BMC Nephrol. 11:312010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gomez-Alamillo C, Benito-Hernandez A,
Ramos-Barron MA, et al: Analysis of urinary gene expression of
epithelial-mesenchymal transition markers in kidney transplant
recipients. Transplant Proc. 42:2886–2888. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loureiro J, Schilte M, Aguilera A, et al:
BMP-7 blocks mesenchymal conversion of mesothelial cells and
prevents peritoneal damage induced by dialysis fluid exposure.
Nephrol Dial Transplant. 25:1098–1108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi SS, Omenetti A, Witek RP, et al:
Hedgehog pathway activation and epithelial-to-mesenchymal
transitions during myofibroblastic transformation of rat hepatic
cells in culture and cirrhosis. Am J Physiol Gastrointest Liver
Physiol. 297:G1093–G1106. 2009. View Article : Google Scholar
|
|
15
|
Xu Y, Wan J, Jiang D and Wu X: BMP-7
counteracts TGF-beta1-induced epithelial-to-mesenchymal transition
in human renal proximal tubular epithelial cells. J Nephrol.
22:403–410. 2009.PubMed/NCBI
|