Introduction
Echinococcus multilocularis (Em) is a
parasite that is a member of the order Cyclophyllidea, the class
Cestoda and the phylum Platyhelminthes. Foxes act as definitive
hosts in the life cycle of Em, whilst small mammals, and, in
particular, rodents (e.g. mice), act as the intermediate hosts.
However, humans may become infected and act as aberrant hosts
through the ingestion of Em eggs. Following the oral uptake of the
eggs, larvae are released from the eggs, and transported through
the blood and lymph vessels to other organs, where they develop
into metacestodes (also known as an alveolar hydatid). Alveolar
echinococcosis (AE) (1), or
alveolar hydatid disease, is a zoonotic helminthic disease caused
by infection with Em larvae. AE is endemic, and is mainly confined
to cold regions of high latitude or the tundra of the Northern
Hemisphere, principally in the continents of North America, Europe
and Asia (2). In China, AE
infections are highly endemic over large areas of northwestern
regions, such as Xinjiang and Gansu, as well as southwestern areas,
such as the Ganzi Tibetan Autonomous Prefecture of Sichuan Province
and Tibet (3,4).
Echinococcosis is severely harmful to human health,
the development of the social economy and animal husbandry
(5). It has a high ten-year
mortality rate, and is regarded as one of the most lethal
helminthic infections (6–9). The metacestode stage of Em in
aberrant hosts is characterized by an alveolar structure, composed
of multiple small vesicles. A typical feature of this stage is its
multi-vesicular budding-like invasive growth, which leads to the
infiltration of the affected organs. Alveolar hydatid disease is
primarily apparent in the liver. During the initial phase, the
damage to hepatic tissue that occurs as a result of alveolar
hydatid infiltration includes mechanical extrusion,
lipopolysaccharide (LPS) stimuli and the direct erosion of the
liver. During the advanced phase, the characteristic symptoms are
liver failure, hepatic coma and portal hypertension, complicated by
gastrointestinal bleeding, which may be fatal (10,11).
In addition to the liver, alveolar hydatid infection may be
transferred through the lymphatic or blood circulation to the lungs
and brain, as well as to the surface and parenchyma of other organs
and the body cavity, in the manner of a malignant tumor.
The initial stages of Em larvae infection are
asymptomatic, and, consequently, the diagnosis of AE only occurs
incidentally, when patients undergo B-mode ultrasound screening of
the liver. Therefore, AE is usually diagnosed at an advanced phase.
Furthermore, in >50% of the patients who are admitted for
treatment, the infection is too advanced to be treated surgically
(12). At present, there are
several options for the treatment of AE; surgery in combination
with chemotherapy remains the first choice therapy. However,
surgical resection is often incomplete, due to the diffuse
infiltration of nonresectable structures. Certain procedures have
been established to prevent the disease, including the regular
deworming of dogs and public education; however, these preventative
measures, as well as the treatment methods, are ineffective. Thus,
there is a requirement to develop preventative and treatment
measures that have an improved efficacy.
A previous study revealed that immunizing the
intermediate hosts with an Echinococcus granulosus (Eg)
recombinant protein vaccine protected against oncosphere infection,
with an immune protective effect of up to 95–100%. Therefore,
immune prevention may be an effective measure that could be taken
to prevent the epidemic of hydatid disease (4). Epitopes, also known as antigenic
determinants, are chemical groups that determine the specificity of
the antigen. Epitopes comprise the fundamental structural subunits
of the T- and B-cell antigen receptors (TCR and BCR, respectively),
and the specific antibody-binding sites. Thus, epitopes may be
classified as T- or B-cell epitopes (13,14).
With the development of bioinformatic technology, epitope vaccines
(15) have become increasingly
important in immune prevention. Superior to traditional vaccines,
vaccines based on epitopes are currently used as protective
measures against infectious diseases. A major challenge in the
development of epitope-based vaccines is to establish the
immunogenic sites of the antigen that exhibit the greatest efficacy
(16). Pfaff et al
(17) demonstrated that the
146–154 amino acid (aa) and the 200–213aa peptides in the
foot-and-mouth disease virus (FMDV) contained immunological
epitopes for the neutralization of the virus, and that it was
possible to use these epitopes for the preparation of epitope
vaccines. In addition, Kouguchi et al (18) revealed that the Emy162 recombinant
antigen induced a 74.3% protective immune effect in rats. In order
to protect against the larval-stage infection of Em, Katoh et
al (19) cloned the Em95
antigen, and generated a vaccine based on this antigen. Compared
with the control group, the protection efficiency in the group
immunized with the recombinant Em95 vaccine was 78.5–82.9%. These
results suggested that the prevention of hydatid disease by a
molecular vaccine is feasible. In the present study, we used
computer technology and molecular biology software to predict and
analyze the secondary and tertiary structures, and the T- and
B-cell epitopes of Emy162, based on the Emy162 gene sequences. The
results revealed the dominant epitopes of the Emy162 antigen, and
provided experimental data for the preparation of an epitope
vaccine.
Materials and methods
Amino acid sequence of the Emy162
protein
The nucleotide sequence of Emy162 was determined
using GenBank (GenBank no. AB303298.1; http://www.ncbi.nih.gov/genbank/). According to
GenBank, the Emy162 protein is composed of 153 amino acid residues,
encoded by the 5–466 region of the Emy162 mRNA. The amino acid
sequence of the Emy162 protein is displayed in Fig. 1.
Prediction of the secondary structure of
the Emy162 protein
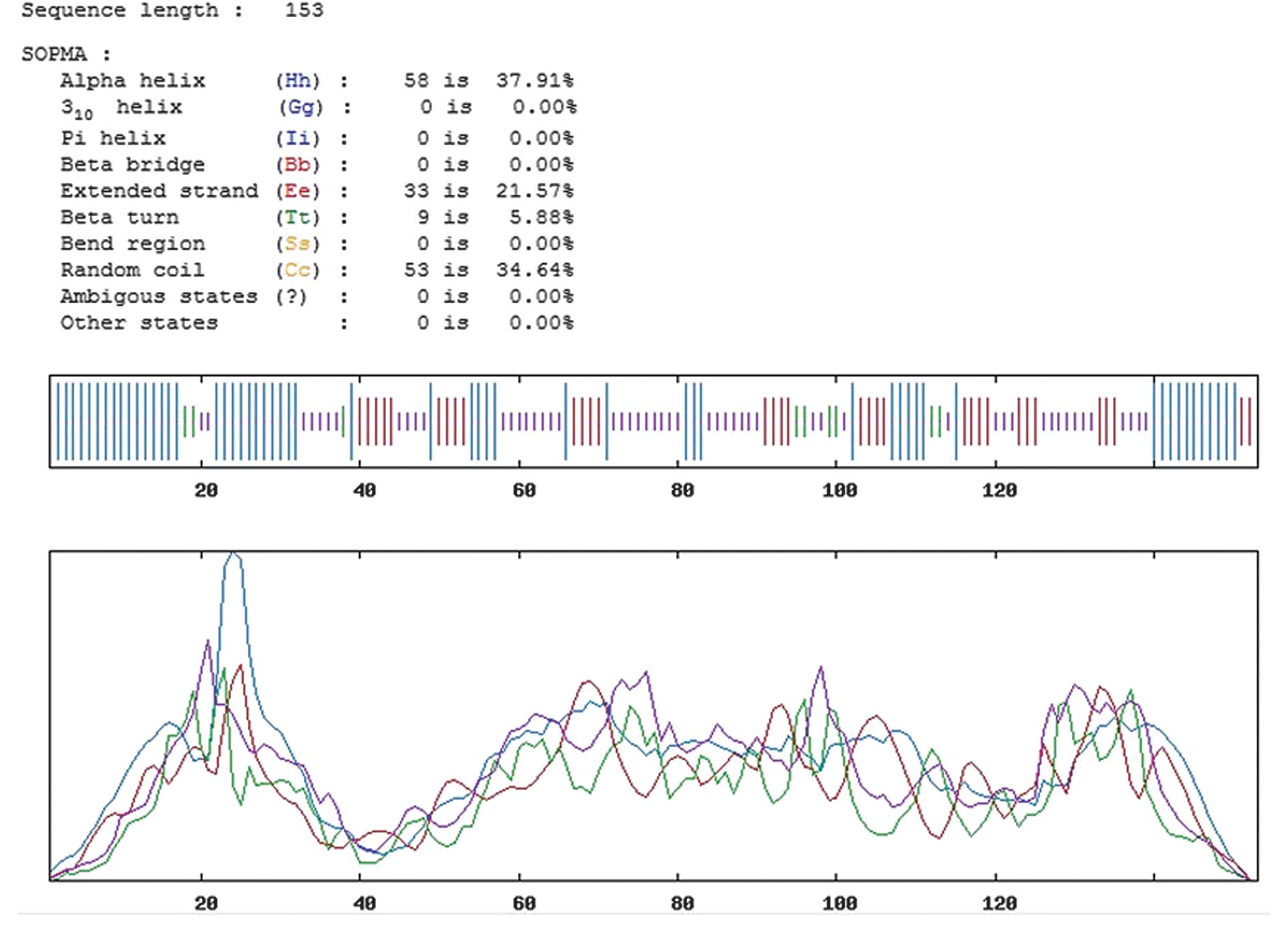
The secondary structure of the Emy162 protein was
predicted by the improved self-optimized prediction method (SOPMA)
software (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html)
(20). The protein sequence of the
Emy162 protein was input, and four conformational states, including
helices, sheets, turns and coils, were analyzed. The parameters of
similarity threshold and window width were set to 8 and 17,
respectively, whilst the remaining parameters were not
adjusted.
Prediction of the T-cell epitopes for the
Emy162 antigen
The major histocompatibility complex (MHC)-I human
leukocyte antigen (HLA)-A*0201-restricted T-cell epitopes were
predicted using online prediction software from the Immune Epitope
Database (IEDB; http://tools.immuneepitope.org/main/index.html)
(21) and Syfpeithi (http://www.syfpeithi.de). The protein sequence of the
Emy162 protein was input, and the parameters were adjusted so that
‘MHC allele(s)’ was set at HLA-A*02:01, and ‘length’ was set at 8,
9 and 10. The remaining parameters were not altered.
Prediction of the B-cell epitopes for the
Emy162 antigen
The B-cell epitopes of the Emy162 protein were
predicted using Bcepred (http://www.imtech.res.in/raghava/bcepred/bcepred_submission.html)
and ABCpred (http://www.imtech.res.in/raghava/abcpred/) online
software from the Institute of Microbial technology, India (Imtech)
(22). The amino acid sequence of
the Emy162 protein was input, and then the parameters were adjusted
to the following values: Hydrophilicity, 2; flexibility, 1.9;
exposed surface area, 2.4; and antigenic propensity, 1.8. The
remaining parameters were not altered.
Prediction of the tertiary structure of
the Emy162 protein
Predictive analysis of the Emy162 protein tertiary
structure was conducted using 3DLigandSite, the online
ligand-binding site prediction server (http://www.sbg.bio.ic.ac.uk) (23). This web server automates the manual
processes used for the prediction of ligand-binding sites in the
eighth round of the critical assessment of protein structure
prediction (CASP8) (24), and is a
useful tool for the analysis of protein tertiary structure. The
site uses ligands from similar structures to make predictions, as
well as providing details of conservation information. Following
the use of 3DLigandSite, the three-dimensional structure of the
protein was analyzed by the Vector Alignment Search Tool (VAST;
http://www.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html).
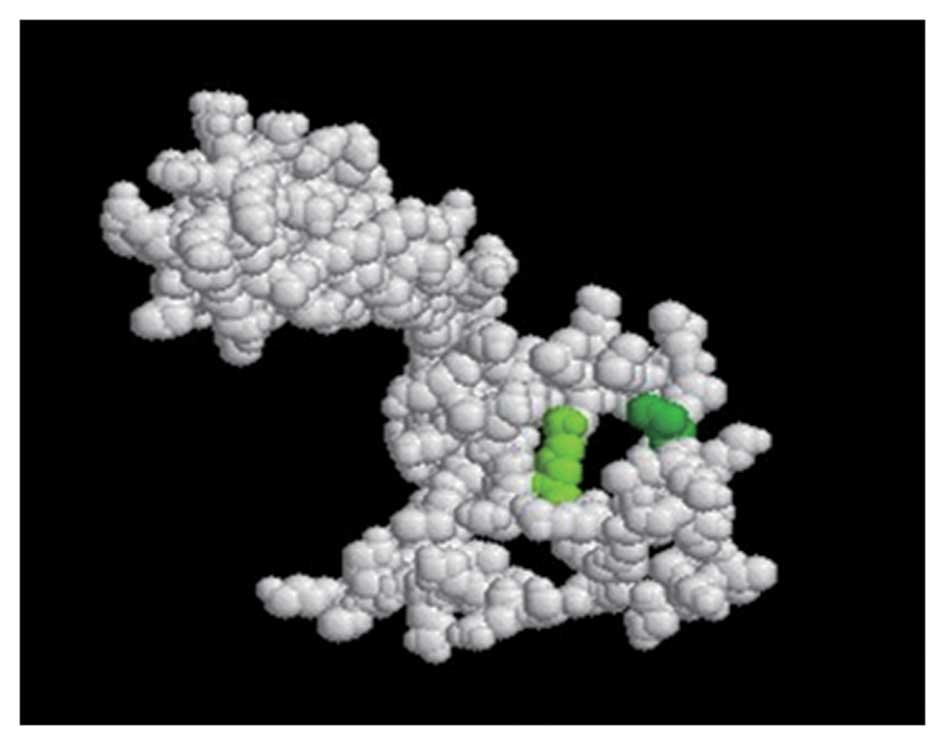
The true epitopes of the structural surfaces were displayed as
green and olive green spheres, whilst the white spheres represented
the remainder of the protein.
Results
Prediction of the secondary structure of
the Emy162 protein
In order to assess the antigenic features of the
Emy162 protein, we predicted its secondary structure using SOPMA
Server software. A greater proportion of extended strands and
random coils present in the structure of the protein corresponded
with an increased likelihood of the protein forming an antigenic
epitope. The predicted secondary structure results for the Emy162
protein are demonstrated in Fig.
2. The results revealed that the proportion of random coils, β
turns, α helices and extended strands (β folds) accounted for
34.64, 5.88, 37.91 and 21.57% of the secondary structure,
respectively.
Prediction of the T-cell epitopes for the
Emy162 antigen
As mentioned previously, there are two types of
epitopes: T- and B-cell epitopes. In order to develop an epitope
vaccine, it is essential to determine the precise location of the
epitope. In the current study, the MHC I HLA-A*0201-restricted T
-cell epitopes were predicted using two online prediction software
applications, IEDB and Syfpeithi, which represented the probability
of a particular region forming a T-cell epitope by a score. The
higher the score assigned to the region, the greater the likelihood
of that region forming antigenic epitopes. The 11 regions that were
revealed to score highly are listed in Table I. It is worthy of note that the two
software solutions utilized different scoring systems. As predicted
by the IEDB software, the high scores ranged between 87.1 and
96.05. However, the high scores predicted by the Syfpeithi software
ranged between 19 and 26. Despite the difference, these
high-scoring regions all had strong potential as epitope
regions.
 | Table I.Analysis of the MHC I
HLA-A*0201-restricted T-cell epitopes using IEDB and Syfpeithi
online prediction software. |
Table I.
Analysis of the MHC I
HLA-A*0201-restricted T-cell epitopes using IEDB and Syfpeithi
online prediction software.
| No. | IEDB | Syfpeithi |
|---|
|
|
|---|
| Starting point | Amino acid
sequence | Score | Starting point | Amino acid
sequence | Score |
|---|
| 1 | 16 | AEEVGVDPE | 96.05 | 39 | LIAKLTKKL | 26 |
| 2 | 23 | PELIAKLTK | 87.30 | 141 | ALIFAMAGL | 26 |
| 3 | 26 | IAKLTKKLQ | 93.00 | 6 | CLILIATSV | 25 |
| 4 | 30 | TKKLQTTLP | 90.45 | 2 | VLRFCLILI | 24 |
| 5 | 31 | KKLQTTLPE | 92.45 | 7 | LILIATSVI | 22 |
| 6 | 82 | RYRNVPIER | 90.20 | 21 | VDPELIAKL | 22 |
| 7 | 84 | RNVPIERQK | 95.30 | 13 | SVIAEEVGV | 21 |
| 8 | 88 | IERQKLTLE | 87.10 | 126 | TLAPGEDGA | 21 |
| 9 | 97 | GLKPSTFYE | 89.10 | 36 | TLPEHFRWI | 20 |
| 10 | 117 | VYKYTGFIR | 91.95 | 94 | TLEGLKPST | 19 |
| 11 | 128 | APGEDGADR | 87.90 | 119 | KYTGFIRTL | 19 |
The results from the IEDB software indicated that
the T-cell epitopes were located at positions 16–26, 30–39, 82–95,
97–105, 117–125 and 128–136, whereas the Syfpeithi software
predicted the epitopes to be located at positions 2–19, 21–29,
36–47, 94–103, 119–135 and 141–149. The five highest-scoring
regions, selected from the combined results of the two software
applications, were 16–29, 36–39, 97–103, 119–125 and 128–135.
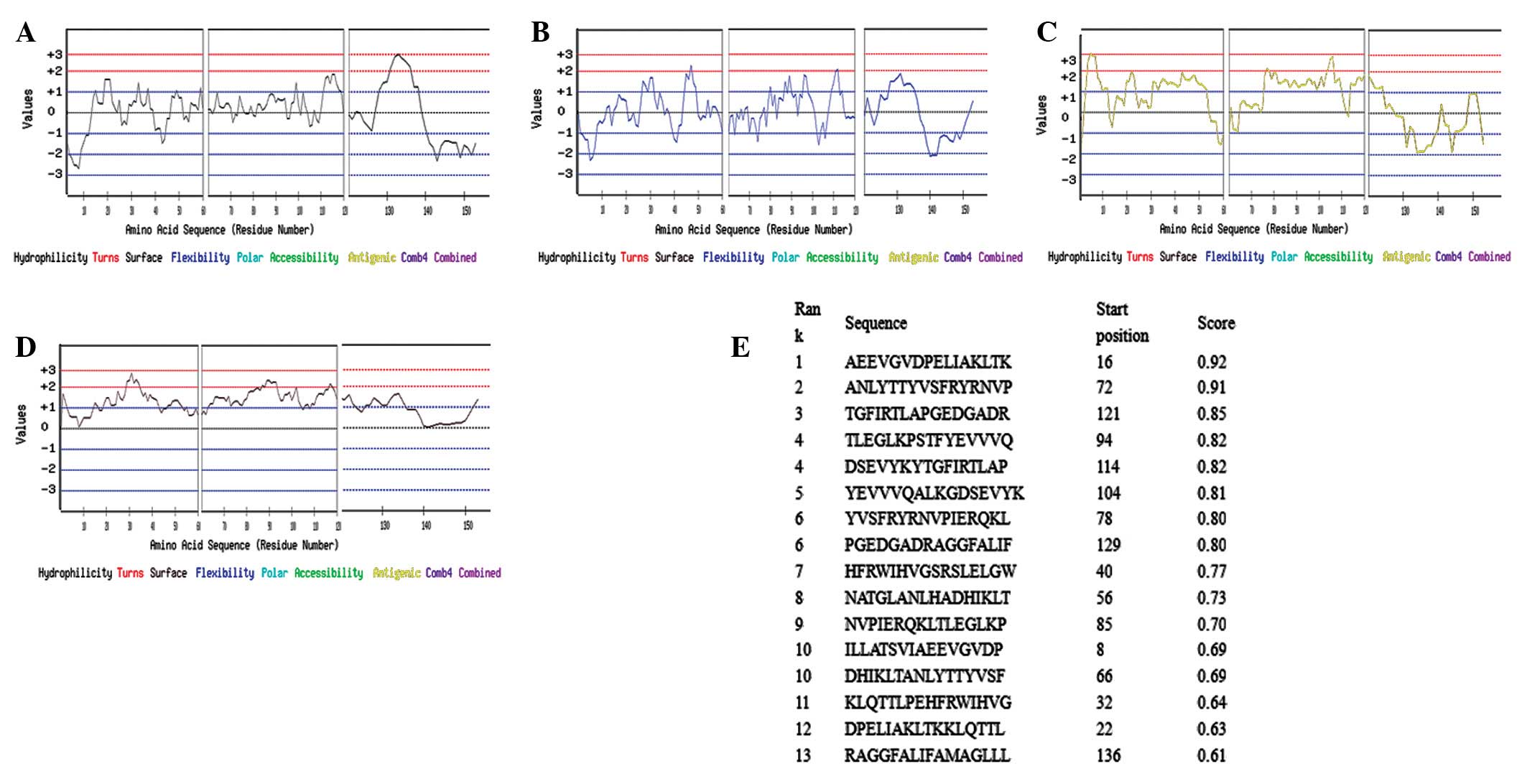
Prediction of the B-cell epitopes for the
Emy162 antigen
The B-cell epitopes were predicted using the Bcepred
online software from Imtech. Based on the features of this
software, the amino acid sequence was divided into three segments
prior to the analysis, and then the parameters of hydrophilicity,
flexibility, the antigenic propensity and the exposed surface area
of antigen were analyzed. The results are displayed in Fig. 3. The region that was revealed to
have high hydrophilicity was 128–139 (amino acid sequence:
APGEDGADRAGG) (Fig. 3A), whilst
the flexible regions were 44–55 (IHVGSRS) and 109–115 (QALKGDS;
Fig. 3B). The possible antigenic
regions were demonstrated to be 1–10 (MVLRFCLILL), 19–25 (VGVDPEL),
74–81 (LYTTYVSF) and 101–109 (STFYEVVVQ; Fig. 3C). The exposed surface areas were
located at positions 28–36 (KLTKKLQTT) and 87–93 (PIERQKL; Fig. 3D).
To further verify these results, the prediction was
also conducted using an additional online software application,
ABCpred. As indicated in Fig. 3E,
the regions with high scores were 16–31, 40–54, 56–70, 72–100,
104–112 and 114–136.
A combination of the results predicted by the
different methods indicated that the potential B-cell epitopes of
the Emy162 antigen were located at positions 8–10, 19–25, 44–50,
74–81, 87–93, 104–109 and 128–136.
Prediction of the tertiary structure of
the Emy162 protein
The tertiary structure of the Emy162 protein was
obtained using the 3DLigandSite software, and compared with the
structure from VAST. The results of the predicted conformations of
the epitopes are displayed in Fig.
4. The olive green and the green spheres indicate potential
epitopes.
Discussion
The research and development of epitope vaccines is
a difficult and highly targeted technology, which comprehensively
utilizes molecular biology and immunology. A key step in the
preparation of the vaccines is obtaining the necessary information
concerning the epitope. In recent years, with the development of
bioinformatics, epitope prediction has improved in simplicity and
significance. Performing predictions with a multi-parameter and
-method analysis greatly enhances the accuracy of the epitope
prediction. In a study by Shen et al (25) the secondary structure and surface
characteristics of the follicle stimulating hormone receptor (FSHR)
extracellular domain were analyzed by DNAStar Protean software, and
the B-cell epitope prediction was conducted using alternative
online software. The possible antigenic epitopes of the FSHR
extracellular domain were predicted, the peptides of the epitopes
were synthesized and then the immunogenicity of these peptides was
determined. In a different study, conducted by Li et al
(26), the B-cell dominant
epitopes of the Epstein-Barr nuclear antigen (EBNA)-1 were
predicted, based on its gene sequences, using SOPMA, as well as the
Garnier-Osguthorpe-Robson (GOR) and hierarchical neural network
(HNN) methods. In addition, the transmembrane domain and various
parameters, including hydrophilicity, were analyzed. Following
this, the sequence alignment of the predicted EBNA-1 B-cell
epitopes and the sequences of interrelated human autoantigens were
contrastively analyzed using blastp. The secondary structure of a
protein is closely correlated with its epitope distribution.
Hydrophilicity and antigenicity are the primary factors involved in
epitope formation, although interrelated factors, such as
flexibility, the exposed surface area and the conformation of the
secondary structure, are also important. Thus, we analyzed the
secondary structure of the Emy162 protein in order to obtain the
antigenic features of the protein. However, as the combined effects
of T- and B-cells are necessary for the elimination of antigens, it
was also important to analyze the T- and B-cell epitopes of the
Emy162 antigen. Therefore, we predicted the T- and B-cell epitopes
using a multi-parameter and -method analysis. A comprehensive
analysis of this nature may, in the future, improve the accuracy
and specificity of epitope prediction.
The α helices and β sheets, in the secondary
structure of proteins, are very regular structures, and are not
readily deformed. This is due to the presence of hydrogen bonds,
which act to maintain structural stability. However, α helices and
β sheets are usually located inside the protein, which is difficult
for ligand binding. By contrast, the β turn and the random coil
regions are located on the surface of the protein, where it is
necessary for the surface structure to make appropriate changes to
meet the functional needs of the protein. Therefore, these
structures are suitable for binding ligands, and have a high
possibility of forming epitopes. As analyzed by SOPMA Server
software, the proportions of α helices and β sheets were 37.91 and
21.57%, respectively. This result indicates that the Emy162 antigen
had a good stability. Random coils and β turns, which represented
the potential epitope regions, accounted for 34.64 and 5.88% of the
protein, respectively. Collectively, there were eight potential
epitope regions in the Emy162 antigen, and, among these eight,
there were high proportions of random coils located at positions
17–28, 61–72, 102–108 and 138–145. This suggested that these four
regions had stronger antigenicity.
The accuracy rate of the MHC I epitope prediction in
the prediction of T-cell epitopes has been demonstrated to be up to
90% (27). In the Chinese
population, HLA-A*0201 is the most common HLA-I molecule, with a
positive rate of 55%. Therefore, in the present study, the
HLA-A*0201-restricted epitopes of the Emy162 antigen were analyzed
using the IEDB and Syfpeithi online software applications. The
T-cell epitopes of the Emy162 antigen were predicted to be located
at positions 16–29, 36–39, 97–103, 119–125 and 128–135, as these
regions exhibited high scores.
In order to improve the accuracy of the B-cell
epitope prediction for the Emy162 antigen, a multi-method and
-parameter analysis was utilized. The hydrophilicity parameter
prediction reflected the position of a hydrophilic residue in the
entire amino acid sequence of the antigen. The amino acid residues
of the protein may be divided into two types: hydrophilic and
hydrophobic residues. Hydrophobic residues are, in general,
packaged inside the protein, whereas hydrophilic residues are
located on the surface of the protein. This conformation is
favorable for the binding of hydrophilic residues to polar
molecules in the solution. Such binding neutralizes the charge of
the protein, and enables the protein to maintain its state of free
minimal energy. Thus, the hydrophilic regions are closely
associated with the epitopes (28). The flexibility parameter prediction
indicated the ability of the protein to bend and fold. With an
increased degree of flexibility, the polypeptide skeleton of the
protein has an improved capacity to fold and bend, thus
facilitating the formation of the secondary structure (29). The antigenic propensity analysis
demonstrated the immunogenic regions of the antigen. Potentially
dominant epitopes are particularly likely to be located in regions
with a high antigenic propensity. The exposed surface area analysis
reflected the distribution of the residues in the outer layer of
the protein (30). The exposed
surface areas of the antigen have an enhanced probability of coming
into contact with the solvent molecules. In combination with these
parameters, two online software applications were used to predict
the B cell epitopes. Seven potential epitope regions were revealed,
located at positions 8–10, 19–25, 44–50, 74–81, 87–93, 104–109 and
128–136.
Protein tertiary structure, one of the higher-order
structures of the protein, is a three-dimensional conformation of
the naturally folded protein. Tertiary structure has a globular
conformation that is formed by the further coiling and folding of
the secondary structure. Therefore, the prediction of the tertiary
structure was a useful supplement to the prediction of the Emy162
antigenic epitopes.
The aim of this study was to obtain the
bioinformatic characteristics of the Emy162 antigen. SOPMA Server
software and the 3DLigandsite were used to predict the secondary
and tertiary structures of the Emy162 antigen, respectively, whilst
a number of online prediction software applications, including
IEDB, Syfpeithi, Bcepred and ABCpred, were used for the T- and
B-cell epitope predictions. The prediction results for the
secondary and tertiary structures suggested that there were
potential epitopes present in the Emy162 antigen. The T- and B-cell
epitope prediction results indicated that the T-cell epitopes were
located at positions 16–29, 36–39, 97–103, 119–125 and 128–135,
whereas the B-cell epitopes were located at positions 8–10, 19–25,
44–50, 74–81, 87–93, 104–109 and 128–136. These regions were the
potential dominant epitopes of the Emy162 antigen. The results of
our study provide experimental data for the identification and
screening of epitopes, and may be used for the development of
epitope vaccines that have an enhanced safety and efficacy. This
may result in the provision of improved regimens for the prevention
and treatment of AE.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (grant nos. 31000411,
81160378, 30901374, 81060135, 81160200 and 30860263), and The Key
Science Project of Xinjiang Autonomous Region XYDXK50780328.
References
|
1.
|
Jenkins EJ, Schurer JM and Gesy KM: Old
problems on a new playing field: Helminth zoonoses transmitted
among dogs, wildlife, and people in a changing northern climate.
Vet Parasitol. 182:54–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Eckert J, Schantz PM, Gasser P, Torgerson
PR, Bessonov AS, Movsessian SO, Thakur A, Grimm F and Nikogossian
MA: Geographic distribution and prevalence. WHO/OIE Manual on
Echinococcosis in Humans and Animals: a Public Health Problem of
Global Concern. Eckert J, Gemmell MA, Meslin F-X and Pawlowski ZS:
World Organisation for Animal Health; Paris: pp. 100–134. 2001
|
|
3.
|
Craig PS; Echinococcosis Working Group in
China: Epidemiology of human alveolar echinococcosis in China.
Parasitol Int. 55(Suppl): S221–S225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Nunnari G, Pinzone MR, Gruttadauria S,
Celesia BM, Madeddu G, Malaguarnera G, Pavone P, Cappellani A and
Cacopardo B: Hepatic echinococcosis: clinical and therapeutic
aspects. World J Gastroenterol. 18:1448–1458. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Rebmann T and Zelicoff A: Vaccination
against influenza: role and limitations in pandemic intervention
plans. Expert Rev Vaccines. 11:1009–1019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Eckert J and Deplazes P: Biological,
epidemiological, and clinical aspects of echinococcosis, a zoonosis
of increasing concern. Clin Microbiol Rev. 17:107–135. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Leggatt GR and McManus DP: Sequence
homology between two immunodiagnostic fusion proteins from
Echinococcus multilocularis. Int J Parasitol. 22:831–833.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ito A: Introduction of ongoing research
projects on echinococcosis at Asahikawa Medical College and some
comments on the surveillance, prevention and control of alveolar
echinococcosis in Japan. Hokkaido Igaku Zasshi. 76:3–8. 2001.(In
Japanese).
|
|
9.
|
Reuter S, Merkle M, Brehm K, Kern P and
Manfras B: Effect of amphotericin B on larval growth of
Echinococcus multilocularis. Antimicrob Agents Chemother.
47:620–625. 2003.PubMed/NCBI
|
|
10.
|
Grosso G, Gruttadauria S, Biondi A,
Marventano S and Mistretta A: Worldwide epidemiology of liver
hydatidosis including the Mediterranean area. World J
Gastroenterol. 18:1425–1437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mandal S and Mandal MD: Human cystic
echinococcosis: epidemiologic, zoonotic, clinical, diagnostic and
therapeutic aspects. Asian Pac J Trop Med. 5:253–260. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kantarci M, Bayraktutan U, Karabulut N,
Aydinli B, Ogul H, Yuce I, Calik M, Eren S, Atamanalp SS and Oto A:
Alveolar echinococcosis: spectrum of findings at cross-sectional
imaging. Radiographics. 32:2053–2070. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Almeida RR, Rosa DS, Ribeiro SP, Santana
VC, Kallás EG, Sidney J, Sette A, Kalil J and Cunha-Neto E: Broad
and cross-clade CD4(+) T-cell responses elicited by a DNA vaccine
encoding highly conserved and promiscuous HIV-1 M-group consensus
peptides. PloS One. 7:e452672012.
|
|
14.
|
Zhang Z, Chen J, Shi H, Chen X, Shi D,
Feng L and Yang B: Identification of a conserved linear B-cell
epitope in the M protein of porcine epidemic diarrhea virus. Virol
J. 9:2252012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ben-Yedidia T and Arnon R: Towards an
epitope-based human vaccine for influenza. Hum Vaccin. 1:95–101.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
You L, Brusic V, Gallagher M and Bodén M:
Using Gaussian process with test rejection to detect T-cell
epitopes in pathogen genomes. IEEE/ACM Trans Comput Biol Bioinform.
7:741–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Pfaff E, Thiel HJ, Beck E, Strohmaier K
and Schaller H: Analysis of neutralizing epitopes on foot-and-mouth
disease virus. J Virol. 62:2033–2040. 1988.PubMed/NCBI
|
|
18.
|
Kouguchi H, Matsumoto J, Yamano K, Katoh
Y, Oku Y, Suzuki T and Yagi K: Echinococcus multilocularis:
purification and characterization of glycoprotein antigens with
serodiagnostic potential for canine infection. Exp Parasitol.
128:50–56. 2011. View Article : Google Scholar
|
|
19.
|
Katoh Y, Kouguchi H, Matsumoto J, Goto A,
Suzuki T, Oku Y and Yagi K: Characterization of emY162 encoding an
immunogenic protein cloned from an adult worm-specific cDNA library
of Echinococcus multilocularis. Biochim Biophys Acta.
1780:1–6. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Geourjon C and Deléage G: SOPMA:
significant improvements in protein secondary structure prediction
by consensus prediction from multiple alignments. Comput Appl
Biosci. 11:681–684. 1995.
|
|
21.
|
Vaughan K, Peters B, Larche M, et al:
Strategies to query and display allergy-derived epitope data from
the immune epitope database. Int Arch Allergy Immunol. 160:334–345.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Sollner J, Grohmann R, Rapberger R, et al:
Analysis and prediction of protective continuous B-cell epitopes on
pathogen proteins. Immunome Res. 4:12008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Wass MN, Kelley LA and Sternberg MJE:
3DLigandSite: predicting ligand-binding sites using similar
structures. Nucleic Acids Res. 38:W469–W473. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Wass MN and Sternberg MJ: Prediction of
ligand binding sites using homologous structures and conservation
at CASP8. Proteins. 77(Suppl 9): 147–151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Shen ZG, Yan P, He W, Chen Z, He H, Zhang
J, Yang X, Wu Y, Liang Z and Li J: Prediction of the secondary
structure and the B cell epitope of the extracellular domain of
FSHR. Journal of Chongqing Medical University. 35:1317–1320.
2010.(In Chinese).
|
|
26.
|
Li L, Zhu S, Li W, Xue X and Zhang L:
Prediction and research on homology of B-cell epitopes of
Epstein-Barr virus nuclear antigen-1. Sheng Wu Yi Xue Gong Cheng
Xue Za Zhi. 28:371–375. 2011.(In Chinese).
|
|
27.
|
Testa JS, Shetty V, Hafner J, Nickens Z,
Kamal S, Sinnathamby G and Philip R: MHC class I-presented T cell
epitopes identified by immunoproteomics analysis are targets for a
cross reactive influenza-specific T cell response. PLoS One.
7:e484842012. View Article : Google Scholar
|
|
28.
|
Zhang W, Liu J, Zhao M and Li Q:
Predicting linear B-cell epitopes by using sequence-derived
structural and physico-chemical features. Int J Data Min Bioinform.
6:557–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Mahdavi M, Mohabatkar H, Keyhanfar M,
Dehkordi AJ and Rabbani M: Linear and conformational B cell epitope
prediction of the HER 2 ECD-subdomain III by in silico methods.
Asian Pac J Cancer Prev. 13:3053–3059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Xue M, Shi X, Zhang J, Zhao Y, Cui H, Hu
S, Gao H, Cui X and Wang YF: Identification of a conserved B-cell
epitope on reticuloendotheliosis virus envelope protein by
screening a phage-displayed random peptide library. PLoS One.
7:e498422012. View Article : Google Scholar : PubMed/NCBI
|