Introduction
Induced pluripotent stem (iPS) cells, generated from
murine somatic fibroblast cells by Takahashi and Yamanaka (1), have revolutionized modern science
(1–9). The iPS cells represent an appealing
option for the derivation of pluripotent patient-specific cells, as
no clinically useful embryos or oocytes are employed, which thereby
avoids ethical obstacles (10).
The original iPS cells were generated by viral transduction of a
limited set of transcription factors (Oct3/4, Sox2, c-Myc and Klf4;
or Oct3/4, Sox2, Lin28 and Nanog), which reprogrammed
somatic cells into pluripotent, embryonic stem cell (ESC)-like
cells (10). Since then, iPS cells
have been generated in different species by a number of methods
(6,8,11–17),
sharing with ESCs the key properties of unlimited self-renewal and
pluripotency. Although human iPS cells may become a more readily
available source of cells for clinical treatment, the question of
whether they are able to maintain their cell self-renewal and
pluripotency during in vitro culture remains. In our
previous studies, we indicated that the expression of numerous
growth factors, including basic fibroblast growth factor (bFGF),
epidermal growth factor (EGF) and insulin-like growth factor 1
(IGF-1), and leukemia inhibitory factor (LIF) by human amniotic
epithelial cells (HuAECs) may be crucial for the function of feeder
cells in maintaining mouse and human ESCs, as well as mouse
spermatogonial stem cells, in an undifferentiated, proliferative
state, capable of self-renewal (18–21).
Furthermore, we have demonstrated that HuAEC-dependent epigenetic
modifications of the c-Myc gene locus occur in the
previously mentioned stem cells, providing a possible mechanism for
their HuAEC-dependent maintenance in an undifferentiated state
(18–20). Although we previously demonstrated
that HuAECs were able to be effectively used as feeder cells, very
little is known about how they maintain iPS cell self-renewal and
inhibit the differentiation of the iPS cells.
In a previous study, Nanog and Oct4
were shown to be two key factors required to maintain the
pluripotency of ESCs, iPS cells and early embryos; they are
co-expressed in developmental stage- and cell type-specific manners
(22). The Nanog gene is
expressed in pluripotent cells, including ESCs, embryonic carcinoma
and embryonic germ cells, and its transcripts are present in the
interior cells of the compacted morula and the inner cell mass of
the blastocyst (22). Oct4
is also necessary for maintaining the pluripotency of cells of
inner cell mass lineage (22), and
its expression has also been observed in ESCs and iPS cells. The
reduction in Oct4 expression leads to trans-differentiation
of ESCs into trophoblast stem cells under adequate culture
conditions (22). Previous studies
have proposed that partial DNA demethylation in restricted areas in
the Oct4 regulatory region is required for gene activation
(3,23–26).
The Nanog promoter is also demethylated in nuclear transfer
ESCs, fibroblast ESCs and in transduced cells (3,23,27).
Moreover, DNA methyltransferase (DNMT)-1 and DNMT3 (a/b) have been
shown to contribute synergistically to the methylation of
Oct4 and Nanog during mouse embryonic cell
differentiation in vivo (28).
Epigenetic regulation, particularly DNA methylation,
is crucial in gene silencing in mammals (28). DNA methylation is important for
establishing the dynamic chromatin configuration of the genome in
pluripotent ESCs and iPS cells and for coordinating genomic
reorganization during cell differentiation (29). A number of key proteins have been
shown to affect epigenetic modifications via DNA methylation, most
importantly the DNA methyltransferases, DNMT1, DNMT3a and DNMT3b
(30). DNMT1 is the ‘maintenance
methyltransferase’ that localizes to replication foci during the S
phase and copies the DNA methylation pattern to the newly
synthesized daughter strand (31,32).
DNMT3a and DNMT3b are de novo methyltransferases,
responsible for the methylation of unmodified DNA (31,32).
Sen et al (33) have
indicated that the DNMT1 protein is predominantly confined to cells
of the basal layer of adult human epidermal tissue and is absent
from the outer differentiated layer. Therefore, DNMT1 is expressed
in epidermal progenitor-containing cell populations and is lost
during differentiation (33).
However, a DNMT1, DNMT3a and DNMT3b triple-knockout ESC line was
shown to grow robustly and maintain its undifferentiated
characteristics (29). In
addition, when ESCs or iPS cells are treated with 5-aza-cytidine (a
DNA methyltransferase inhibitor), the influence of DNMT1 is
weakened and DNA hypomethylation occurs during cell reprogramming
(34). Although DNMT1 is
frequently designated as a maintenance methyltransferase, while
DNMT3a and DNMT3b are classified as de novo
methyltransferases, these enzymes have been shown to exhibit
overlapping functions (29).
Moreover, in spite of a 5-to-30-fold higher preference of DNMT1 for
hemimethylated DNA, it exhibits greater de novo DNA
methyltransferase activity in vitro and is present at higher
levels than DNMT3a and DNMT3b in ESCs and somatic cells (35).
Experimentally, human iPS cells are highly similar
to human ESCs in terms of morphology, proliferation, gene
expression and the epigenetic status of pluripotency-specific genes
(21). Furthermore, the global
epigenetic landscapes, as indicated by the distribution of histone
modifications and DNA methylation, are very similar between ESCs
and iPS cells (29). Therefore,
the cells employ the same molecular mechanisms to maintain the
expression of the pluripotency regulators Nanog and
Oct4 and to maintain their properties via epigenetic
modifications (36). Our
preliminary experiments revealed that HuAECs were able to be
effectively used as feeder cells to maintain iPS cell self-renewal
and inhibit iPS cell differentiation. iPS cells simultaneously
express high levels of Oct4 and Nanog when cultured
on HuAECs. Accordingly, we hypothesized that the low endogenous
activity of DNMT1, DNMT3a and/or DNMT3b in human iPS cells may lead
to hypomethylation of the CpG islands on the promoter regions of
Nanog and Oct4 and that the high expression of these
factors, modulated by HuAECs feeder layers, may maintain the
pluripotency and self-renewal properties of the iPS cells.
Materials and methods
Preparation of mouse embryonic
fibroblasts (MEFs) and HuAECs
MEF cells were isolated from 13-day-old C57BL/6
mouse embryos. Cells were mitotically inactivated using mitomycin C
(Sigma-Aldrich, St. Louis, MO, USA), as described previously
(18). The mitotic inactivation of
MEF cells was conducted by treatment with 10 μg/ml mitomycin
C for 2 h at 37°C. The cells were washed three times with
phosphate-buffered saline (PBS), digested with 0.25% trypsin-EDTA
solution (cat no. 25300-054; Invitrogen Life Technologies,
Carlsbad, CA, USA) and plated at a density of 1×105/ml,
with 2.5 ml in each well of a gelatin-coated six-well dish. Human
placentas were obtained with written and informed consent from
pregnant females who were negative for human immunodeficiency virus
(HIV)-I, hepatitis B and hepatitis C. The study received approval
for the appropriate use of human amnion by the institutional Ethics
Committee of Shanghai Geriatric Institute of Chinese Medicine
(Shanghai, China). Amniotic membranes were mechanically separated
from the chorions of placentas, which were obtained from females
who had undergone an uncomplicated Cesarean section. HuAECs were
harvested from the epithelial layers (with the basement membrane
attached) of the obtained amniotic membranes, as described in a
previous study, with certain modifications (18). In brief, the membrane was placed in
a 250-ml flask containing Dulbecco’s modified Eagle’s medium (DMEM)
and cut with a razor to produce 0.5–1.0 cm2 segments.
The segments were subsequently digested with 0.25% trypsin-EDTA at
37°C for 45 min and the resulting cell suspensions were seeded in a
six-well plate in DMEM supplemented with 10% fetal calf serum (FCS;
PAA Laboratories GmbH, Pasching, Austria), penicillin (100 U/ml)
and glutamine (0.3 mg/ml). Following this, the cells were incubated
in a humidified tissue culture incubator containing 5%
CO2 at 37°C. The HuAECs were grown to a density of ~100%
and were subsequently used as feeder layers for human iPS cell
culture following mitomycin C (Sigma-Aldrich) treatment.
Co-culture of human iPS cells with HuAECs
and MEFs
The human iPS cells were generated by our laboratory
and derived from CD34+ human amniotic fluid cells
(HuAFCs) via transduction with lentiviral constructs encoding only
Oct4, as previously described (37). iPS cultures were separated from the
feeder cells by treatment with 0.125% trypsin-EDTA solution and
plated onto and co-cultured with HuAECs or MEFs. The cells were
cultured in DMEM:F12 (1:1) medium supplemented with 15% KnockOut™
Serum Replacement (Invitrogen Life Technologies), 1 mM sodium
pyruvate, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM
β-mercaptoethanol, penicillin (25 U/ml)-streptomycin (925 mg/ml),
and mixed, without LIF. The cells were incubated in a humidified
tissue culture incubator containing 5% CO2 at 37°C. All
cells were cultured on the same feeder until the 10th passage,
prior to being used for subsequent experiments.
Alkaline phosphatase (AP) staining
The AP activity of human iPS cells, which were
cultured on HuAECs or MEFs, was determined using an alkaline
phosphatase detection kit (Sigma-Aldrich), in accordance with the
manufacturer’s instructions (38).
RNA extraction and analysis using
quantitative polymerase chain reaction (qPCR)
Total-RNA from each cell was isolated using TRIzol
reagent® (Invitrogen Life Technologies), in accordance
with the manufacturer’s instructions. The RNA samples were treated
with DNase I (Sigma-Aldrich), quantified and reverse-transcribed
into complementary DNA (cDNA) with the ReverTra Ace-α First Strand
cDNA Synthesis kit [Toyobo (Shanghai) Biotech Co., Ltd., Shanghai,
China]. qPCR was conducted with a RealPlex4 real-time PCR detection
system from Eppendorf (Hamburg, Germany), with SYBR®
Green Real-Time PCR Master Mix [Toyobo (Shanghai) Biotech Co.,
Ltd.] as the detection dye. The qPCR amplification was performed
over 40 cycles with denaturation at 95°C for 15 sec and annealing
at 58°C for 45 sec. The target cDNA was quantified using the
relative quantification method. A comparative threshold cycle (Ct)
was used to determine gene expression relative to a control
(calibrator); steady-state mRNA levels are expressed as an n-fold
difference relative to the calibrator. For each sample, the maker
gene Ct values were normalized with the formula: ΔCt=Ct_genes -
Ct_18S RNA. To evaluate the relative expression levels, the
following formula was used: ΔΔCt=ΔCt_all_groups -
ΔCt_blank_control_group. The values used to the plot relative
expression of the markers were calculated using the expression
2−ΔΔCt method. The mRNA levels were calibrated on the
basis of levels of 18S ribosomal RNA (rRNA). The cDNA of each gene
was amplified with primers as previously described (21,37,39,40).
RNA interference and transfection
The small interfering RNA (siRNA)-DNMT1 plasmid was
manufactured by Shanghai GenePharma, Ltd. (Shanghai, China) and the
methods used for plasmid transfection were in accordance with the
company’s instructions. In brief, iPS cells were cotransfected with
0.3 μg siRNA-DNMT1 expression plasmid or siRNA-Mock plasmid,
respectively, using Lipofectamine 2000 reagent (Invitrogen, Life
Technologies Corporation, Grand Island, NY, USA), in accordance
with the manufacturer’s instructions. The cells were seeded in a
six-well plate and cultured in DMEM:F12 (1:1) medium supplemented
with 15% KnockOut™ Serum Replacement, 1 mM sodium pyruvate, 2 mM
L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM
β-mercaptoethanol, penicillin (25 U/ml)-streptomycin (925 mg/ml)
and mixed, without LIF. The cells were incubated in a humidified
tissue culture incubator containing 5% CO2 at 37°C until
80% confluence was achieved.
Flow cytometric (FCM) analysis of cell
cycle by propidium iodide (PI) staining
Each group of cells was seeded at a density of
3×105 cells per well in six-well plates and cultured
until 85% confluent. Following this, each group of cells was washed
three times with PBS, prior to being subjected to centrifugation
(Allegra X-22®; Beckman Coulter, Miami, FL, USA) at
1,000 × g for 5 min. The cell pellets were then resuspended in 1 ml
PBS, fixed in 70% ice-cold ethanol and stored in a freezer for
>48 h. Prior to FCM analysis, the fixed cells were centrifuged,
washed twice with PBS and resuspended in PI staining solution
(Sigma-Aldrich) containing 50 μl/ml PI and 250 μg/ml
RNase A (Sigma-Aldrich). The cell suspensions, which were hidden
from light, were incubated for 30 min at 4°C and analyzed using a
fluorescence-activated cell sorter (FACS; FCM-500; Beckman
Coulter). A total of 20,000 events were acquired for analysis using
CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA).
Bisulfite conversion of genomic DNA and
methylation-specific PCR (MS-PCR)
The cells were lysed in DNA lysis buffer [0.5%
sodium dodecyl sulfate (SDS), 0.1 M EDTA, 10 mM Tris-HCl (pH 8.0)
and 100 ng/ml proteinase K; all from Sigma-Aldrich] and incubated
at 55°C for 2 h. The treatment of genomic DNA and the MS-PCR assay
were performed as previously described (2,5,9,18,37).
In addition, the specific primers for Nanog and Oct-4
were designed as previously described (2,5,9,18,37).
The PCR products were separated using 12 g/l ethidium bromide
containing agarose gel electrophoresis with 1X Tris acetate EDTA
(TAE) buffer, and visualized under UV illumination.
Chromatin immunoprecipitation (ChIP)
assays
ChIP experiments were conducted using
anti-acetylated histone H3 antibody (Upstate Biotechnology, Inc.,
Lake Placid, NY, USA), anti-trimethylated H3K27 antibody (Abcam,
Cambridge, UK) and normal rabbit immunoglobulin G (IgG; Upstate
Biotechnology, Inc.) as a negative control. All steps were
performed as previously described (2,5,9,18,37).
The cells were fixed using 1% formaldehyde for 30 min at 37°C and
then quenched using 125 mM glycine for 10 min at room temperature
to form DNA-protein cross-links. Following this, the samples were
placed on ice and sonicated until chromatin fragments became
200–1,000 bp in size. The samples were then incubated with
antibodies at 4°C overnight. The PCR amplification was performed
under the following conditions: 33 cycles of denaturation at 95°C
for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for
30 sec.
Immunofluorescence (IF) staining
The cultured cells were washed three times with FCS
and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min. The
cells were then washed using Tris-buffered saline containing 0.1%
Triton X-100 [TBST-100 buffer; 25 mM Tris-HCl (pH 8.0), 125 mM NaCl
and 0.1% Triton X-100] three times, prior to blocking. Following
blocking, the cells were incubated with rabbit anti-human Oct3/4
polyclonal antibody (1:200; Chemicon, Temecula, CA, USA) and rabbit
anti-human Nanog polyclonal antibody (1:200; Chemicon)
overnight at 4°C, and then with Cy3-conjugated goat anti-rabbit IgG
antibody (1:200; Abcam) and 5 μg/ml
4’,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) at room
temperature for 30 min. Following this, the cells were thoroughly
washed with TBST-100 and viewed under a fluorescence microscope
(DMI3000; Leica Camera Inc., Allendale, NJ, USA).
Western blot analysis
Cells were lysed using a 2X loading lysis buffer [50
mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, 10%
β-mercaptoethanol, 10% glycerol and 0.002% bromphenol blue]. The
total quantity of proteins from the cultured cells was subjected to
12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred
onto Hybrid-polyvinylidene fluoride (PVDF) membranes (Millipore,
Bedford, MA, USA). Following blocking with 5% (w/v) non-fat dried
milk in Tris-buffered saline containing Tween-20 [TBST-20; 25 mM
Tris-HCl (pH 8.0), 125 mM NaCl and 0.05% Tween-20], the PVDF
membranes were washed four times (15 min each) with TBST-20 at room
temperature and incubated with primary antibody. Following
extensive washing, the membranes were incubated with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody
(1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for
1 h. The membranes were then washed four times (15 min each) with
TBST-20 at room temperature, prior to the immunoreactivity being
visualized by enhanced chemiluminescence (ECL) using an ECL
Chemiluminescent Substrate Reagent kit from Perkin-Elmer Life
Science (Norwalk, CT, USA).
Teratoma formation
All animal procedures were conducted at Shanghai
University of Traditional Chinese Medicine (Shanghai, China) with
approval from the Institutional Animal Care and Use Committee and
in accordance with the institutional guidelines. Human iPS cells
(1×106) were inoculated into the hind legs of severe
combined immunodeficient (SCID) mice. Teratomas were embedded in
paraffin and histologically examined following hematoxylin and
eosin staining. The procedure for the teratoma formation experiment
was performed as described previously (18).
Statistical analysis
Each experiment was performed as least three times
and the data are presented as the mean ± standard error (SE). The
differences were evaluated using Student’s t-tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
Pluripotency of iPS cells derived from
CD34+ HuAFCs
We have previously described the successful
generation of iPS cells from CD34+ HuAFCs by
transduction with lentiviral constructs encoding only Oct4
(37). In this study, the iPS
cells were cultured on HuAEC feeder layers until the 10th passage,
prior to use in experiments. After testing the effects of different
feeder layers, the pluripotency of iPS cells was assayed. Under the
microscope, a number of ESC-like colonies were observed among the
feeder cells; these Oct4-green fluorescent protein
(GFP)-positive colonies appeared isolated and rounded, consistent
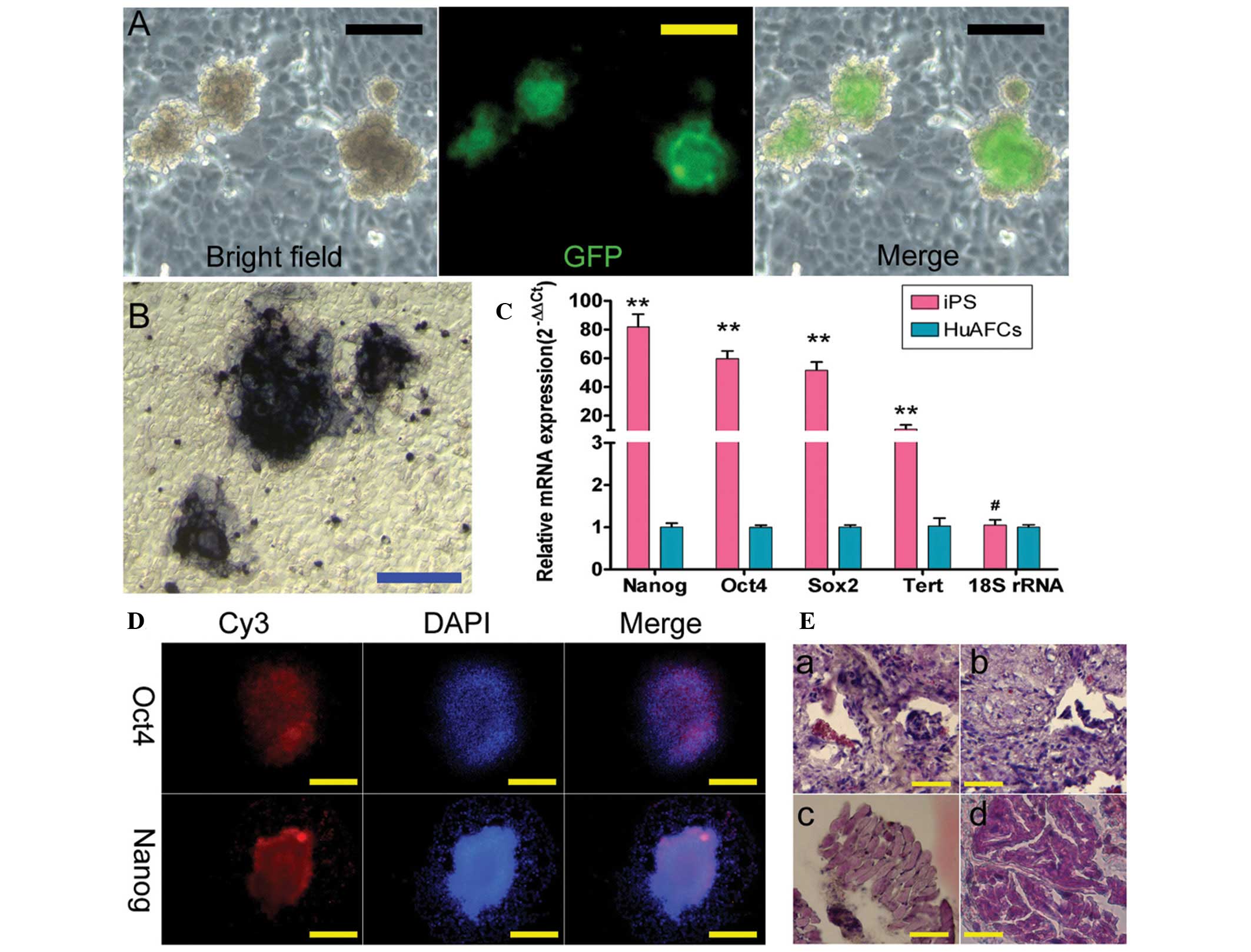
with more undifferentiated cells (Fig.
1A). Furthermore, the AP activity of these human iPS cells was
high, as represented by the deep blue staining on the surface of
the colonies (Fig. 1B). In
addition, the IF staining revealed that the expression levels of
the pluripotent stem cell markers, Nanog, Oct4 and
Sox2, were increased in the iPS colonies (Fig. 1D). Consistent with the IF results,
qPCR analysis showed that the expression levels of these stem cell
markers were ~60–100-fold higher in human iPS cells than in HuAFCs,
which served as an internal control (Fig. 1C). Moreover, the high level of
telomerase activity in human iPS cells suggested that their
replicative life-span was likely to exceed that of somatic cells.
Meanwhile, in the in vivo xenograft experiments, teratomas
formed on the legs of SCID mice injected with the iPS cells
(Fig. 1E). The iPS-derived
teratomas contained cellular representatives of all three germ
layers (Fig. 1E). Based on these
results, it was concluded that the human iPS cells derived from
HuAFCs possessed strong pluripotency.
Pluripotency of iPS cells cultured on
MEFs is weakened during successive subcultures in vitro
iPS cells were successively subcultured either on
HuAECs or MEFs in order to evaluate the effect of the different
feeder layers on pluripotency. The two groups of cells were
cultured in uniform conditions and were subcultured in succession
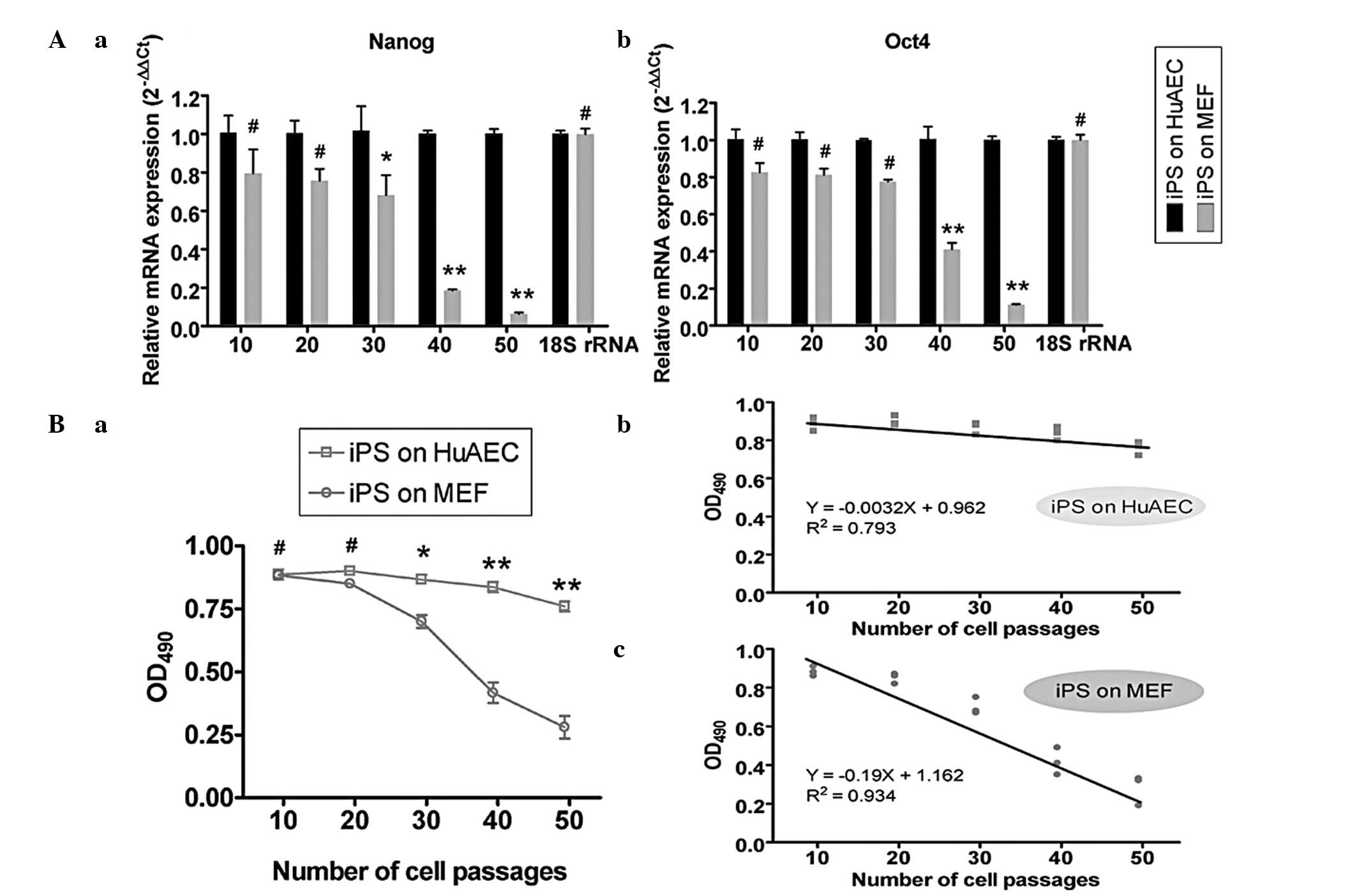
until passage 50. Since AP levels decrease as stem cells lose their
pluripotency and differentiate, the AP activity of human iPS cells
cultured on different feeder layers was analyzed at every 10th
passage. The results showed that the AP activity of the iPS cells
cultured on MEFs reduced rapidly and there was a significant
negative correlation between the AP activity level and the number
of cell passages (R2=0.934, P<0.05). However, when
the iPS cells were cultured on HuAECs, their AP activity level was
not significantly altered (R2=0.793, P>0.05; Fig. 2B). In addition, at the 40th and
50th passage, the AP activity levels of human iPS cells cultured on
MEFs (0.417±0.041 and 0.280±0.045, respectively) were significantly
lower than those on HuAECs (0.837±0.021 and 0.760±0.022,
respectively). To evaluate whether the pluripotency of iPS cells
changed, stem cells markers were assayed using qPCR. The results of
the qPCR analysis showed that the Nanog and Oct4
expression levels in iPS cells cultured on MEFs reduced rapidly,
while those in iPS cells cultured on HuAECs did not markedly change
over time (Fig. 2A).
HuAEC and MEF feeders maintain iPS cells
in the resting stage and early stage of DNA synthesis (G0/G1
stage)
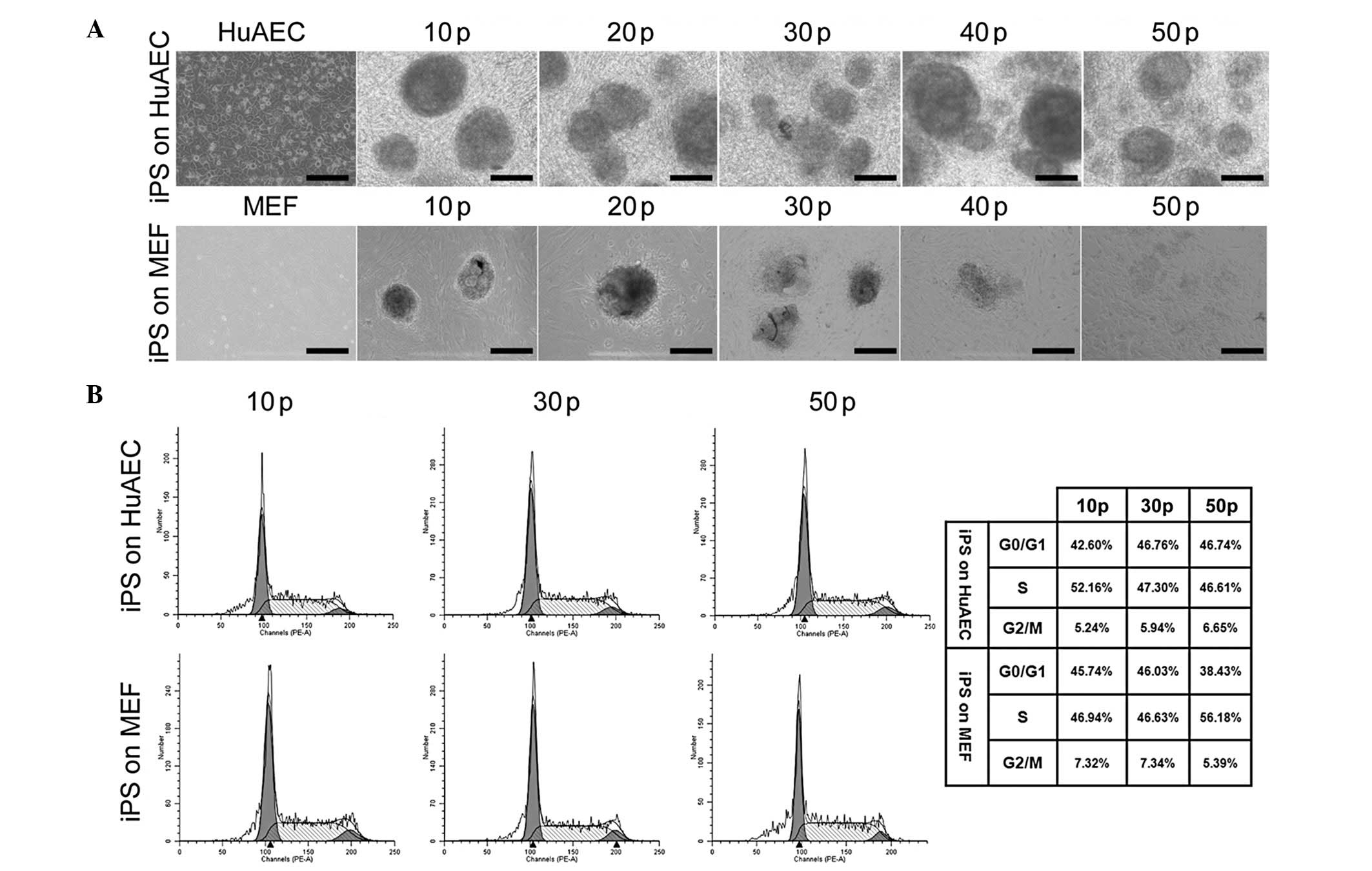
In the early passages (comparing passages 10, 30 and
50), the iPS cells grown on HuAECs and MEFs exhibited similar cell
cycle distributions (Fig. 3).
Furthermore, the cell cycles of the iPS cells cultured on HuAECs
were not markedly different between the 10th, 30th and 50th
passages, indicating that long-term culture on HuAECs feeder layers
did not affect the process of cell division in the iPS cells.
Similarly, when the iPS cells were cultured on MEFs, the FCM
analysis showed no significant differences in the cell cycle
distribution at each passage. The iPS cells cultured on MEFs were
always in the resting stage and early stage of DNA synthesis (G0/G1
stage; Fig. 3). No significant
differences were observed between the cell cycles of the iPS cells
cultured on HuAECs or on MEFs at the 10th, 30th or 50th
passages.
Changes in DNA and histone epigenetic
modifications in iPS cells during consecutive subcultures
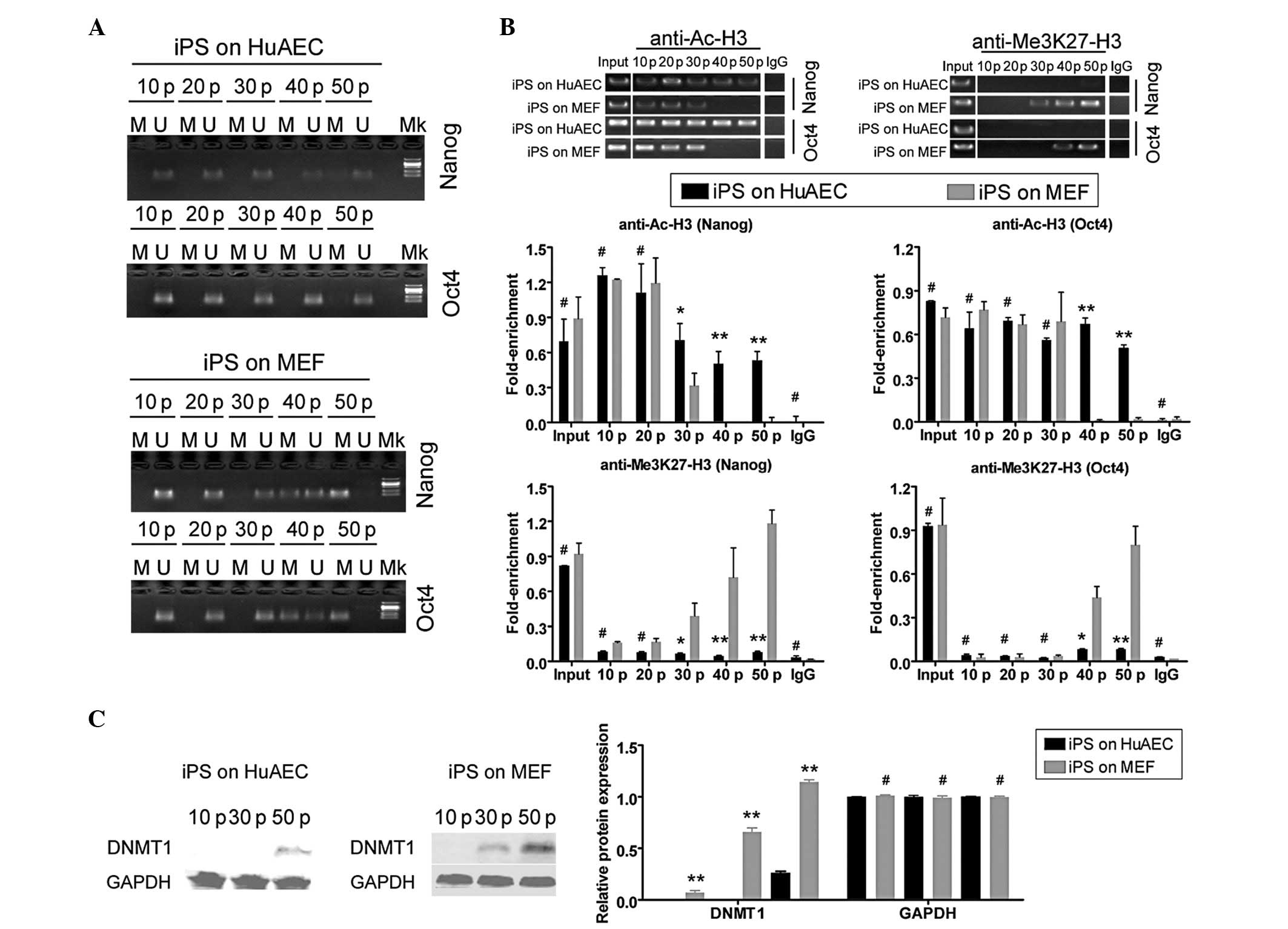
The DNA methylation status of the CpG islands in the
Nanog and Oct4 promoters was investigated using
sodium bisulfite treatment of the iPS cells cultured on HuAECs or
MEFs at every 10th passage. All the CpG islands in the Nanog
and Oct4 promoter regions were demethylated with no
significant differences from the 10th to the 50th passage in human
iPS cells cultured on HuAECs (Fig.
4A). However, in iPS cells cultured on MEFs, these regions of
the Nanog and Oct4 promoters were moderately
demethylated at passages 10, 20 and 30 (Fig. 4A), prior to becoming
hypermethylated at passages 40 and 50. These differences suggested
that HuAECs positively regulated the Nanog and Oct4
genes in human iPS cells by maintaining the hypomethylation of
promoter CpG islands. In addition, ChIP assays were performed to
evaluate the histone H3 acetylation levels of the Nanog and
Oct4 promoters in human iPS cells cultured on HuAECs or
MEFs. In iPS cells cultured on HuAECs, the acetylation and K27
trimethylation of histone H3 in the Nanog and Oct4
promoters and the 5’untranslated regions (5’UTRs) were similar to
those in human iPS cells cultured on MEFs within 20 passages
(Fig. 4B). However, from passage
30, histone H3 appeared relatively hyperacetylated and histone K27
appeared hypomethylated in the Nanog- and
Oct4-specific sites in human iPS cells cultured on HuAECs,
compared with the same regions in cells cultured on MEFs (Fig. 4B). These results suggested that
HuAECs were capable of maintaining the Nanog and Oct4
loci in an active transcriptional state through covalent histone
modifications over long-term culture. Western blotting was also
used to assess the relative protein expression of DNMT1 in iPS
cells cultured on different feeder layers. Over multiple passages,
DNMT1 protein was more highly expressed in iPS cells cultured on
MEFs than in iPS cells cultured on HuAECs (Fig. 4C). At passages 30 and 50, the DNMT1
protein levels in iPS cells cultured on MEFs were 0.676±0.040 and
1.220±0.021 relative to glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) expression levels, respectively. These values were
significantly higher than those in iPS cells cultured on HuAEC
feeders (0.003±0.001 and 0.021±0.010 relative to GAPDH levels,
respectively).
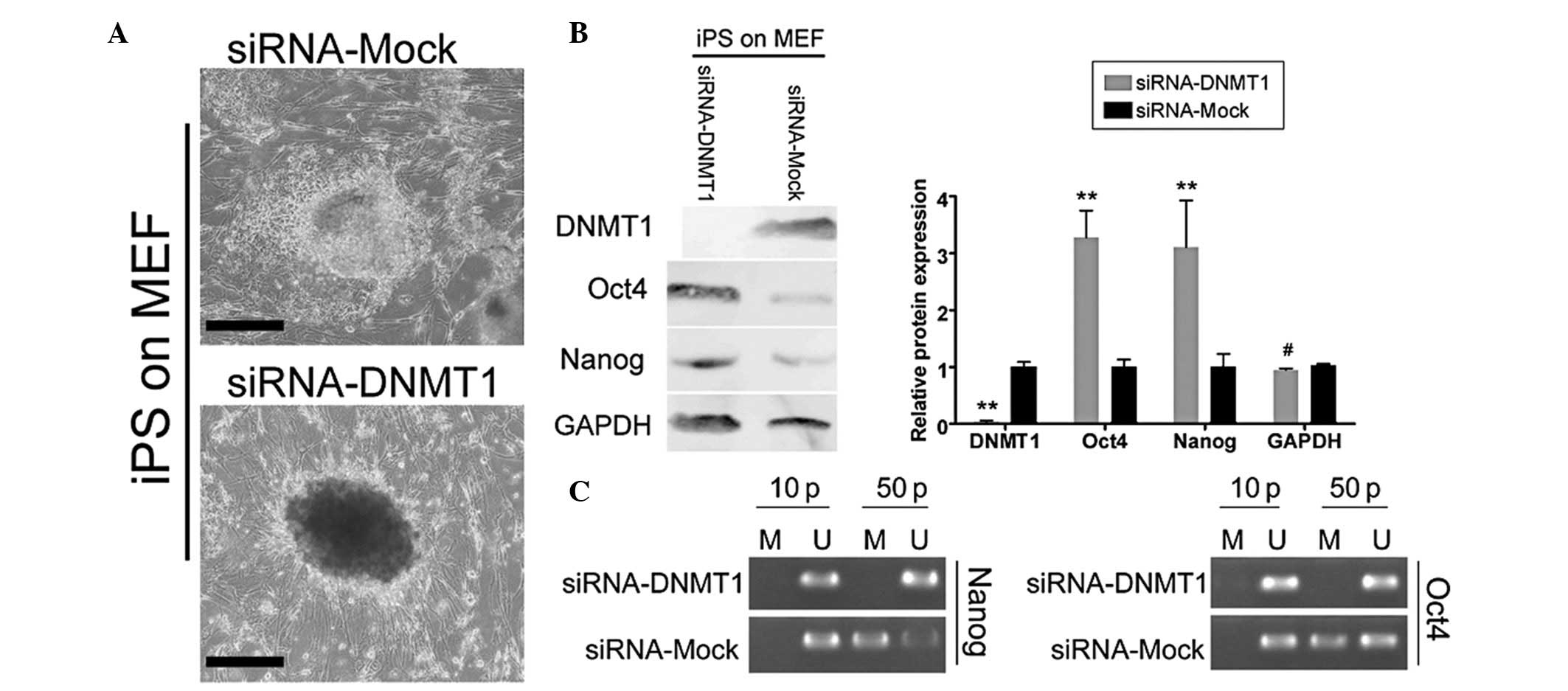
Suppression of endogenous DNMT1
expression in iPS cells maintains their pluripotency while cultured
on MEFs
In order to evaluate whether endogenous DNMT1
expression promoted DNA methylation of Nanog and Oct4
and the differentiation of cultured iPS cells, siRNA-DNMT1 and
siRNA-Mock were transfected into iPS cells. The efficiency of the
transfected siRNA on mRNA and protein expression was assessed using
qPCR and western blotting, respectively, and these experiments were
performed on all cell groups subcultured up to passage 50. As shown
in Fig. 5B, western blotting
indicated that the DNMT1 protein expression in iPS cells
transfected with siRNA-Mock (1.002±0.089) was higher than that in
iPS cells transfected with siRNA-DNMT1 (0.037±0.020, P<0.01,
n=3). These results demonstrated that siRNA-DNMT1 specifically
interfered with DNMT1 expression in iPS cells. Having confirmed
that endogenous DNMT1 expression was suppressed by siRNA, the
pluripotent stem cell biomarkers Oct4 and Nanog in
iPS cells cultured on MEFs were tested using western blotting. The
results revealed that the Oct4 and Nanog protein
levels in the siRNA-DNMT1-transfected iPS cells cultured on MEFs
were 3.277±0.475 and 3.108±0.719 of GAPDH expression levels,
respectively (Fig. 5B). These
values were significantly higher than those in the
siRNA-Mock-transfected iPS cells (1.002±0.128 and 1.005±0.228 of
GAPDH levels, respectively). In addition, the promoter regions of
Nanog and Oct4 were observed to be moderately
hypomethylated or demethylated in siRNA-DNMT1-transfected iPS
cells, while they were moderately hypermethylated in
siRNA-Mock-transfected iPS cells (Fig.
5C). These differences suggested that DNMT1 positively
regulated the Nanog and Oct4 genes in human iPS cells
through the de novo or maintained hypermethylation of the
CpG islands, and that high DNMT1 expression in the iPS cells was
able to promote their differentiation.
Discussion
In the early stages of generation, iPS cells are
generally highly efficient in their ability to be reprogrammed and
possess a strong capacity for self-renewal. However, following
consecutive subculturing, it was observed in this study that iPS
cells began to lose these characteristics and to show decreased
pluripotency as they differentiated, particularly following 40
passages. Moreover, when the iPS cells cultured on MEFs were
subcultured continuously in vitro, the cells also showed a
decreased expression of endogenous pluripotency stem cell markers,
and the ability of the cells to differentiate into three germ
layers in vivo was not as efficient as that of iPS cells
cultured at earlier passages. Furthermore, assays at later passages
indicated that the expression of two crucial transcriptor factors,
Oct4 and Nanog, decreased rapidly during the in
vitro subculture of the iPS cells on MEFs. A previous study
demonstrated that the promoter regions of Oct4 and
Nanog, as well as other pluripotency regulators, were
methylated in somatic cells and became demethylated during
reprogramming to a pluripotent state (29). The downregulation of Nanog
and Oct4 expression may induce the pluripotent stem cells to
differentiate (41). The effects
of DNA methylation on cells include transcriptional repression by
the methylation of promoter regions, and this is required in
mammals for embryonic development, X chromosome inactivation and
imprinting (42). Moreover, a
previous study revealed that three human DNA methyltransferases
(DNMT1, DNMT3a and 3b) were widely expressed in a coordinated
fashion in the majority of normal tissues, tumors and stem cells
(42). In our previous studies, we
have also demonstrated that the expression of a number of growth
factors (bFGF, EGF and IGF-1) and LIF by HuAECs may be crucial
components by which feeder cells maintain mouse and human ESCs, as
well as mouse spermatogonial stem cells, in an undifferentiated,
proliferative state, capable of self-renewal (18,21,37,39,40,43).
With reference to these studies, we hypothesized that HuAECs as
feeder layers may support the undifferentiated growth and maintain
the pluripotency of iPS cells in long-term culture. The two main
conclusions from this study are discussed in the following
section.
DNMT1 was shown to induce hypermethylation of the
promoters of Oct4 and Nanog, although not those of
DNMT3a or 3b, causing the iPS cells to lose
pluripotency during continuous subculture. We analyzed the
expression levels of DNMT1 in iPS cells at different passages. Over
time, the DNMT1 expression in these iPS cells became increasingly
higher. Having confirmed that our siRNA specifically reduced
endogenous DNMT1 expression in iPS cells, it was observed that the
expression of pluripotent stem cell markers (Nanog and
Oct4), as well as the level of AP activity, was higher in
iPS cells transfected with siRNA-DNMT1 than those in iPS cells
transfected with siRNA-Mock. Importantly, compared with the
siRNA-Mock-transfected group, specific loci of Nanog and
Oct4 were demethylated/hypomethylated in the iPS cells
transfected with siRNA-DNMT1 during long-term subculture.
Therefore, we concluded that DNMT1, not DNMT 3a/3b, induced the
hypermethylation of Nanog and Oct4 and caused the iPS
cells to lose pluripotency during long-term subculture.
A further conclusion from this study was that the
use of HuAECs as feeder layers was able to maintain the
pluripotency of human iPS cells during long-term subculture. It was
demonstrated that the HuAEC feeder cells allowed human iPS cells to
maintain a high level of AP activity. In addition, it was observed
that expression levels of Nanog, Oct4 and other
important stem cell markers were higher in iPS cells cultured on
HuAECs compared with those cultured on MEFs during long-term
subculture. Furthermore, using the sodium bisulfite and PCR assay,
it was demonstrated that the CpG islands of the Nanog- and
Oct4-specific loci were hypomethylated in iPS cells cultured
on HuAECs. In addition, iPS cells cultured on HuAECs exhibited
higher levels of histone H3 acetylation and lower levels of H3K27
trimethylation at the Nanog- and Oct4-specific loci
than those cultured on MEFs during subculture. Accordingly, the
expression of DNMT1 in iPS cells cultured on HuAECs was lower than
that in the cells cultured on MEFs. In combination, these results
suggested that the HuAEC-induced epigenetic modifications at the
Nanog and Oct4 loci may be a key mechanism to
maintain the iPS cells in an undifferentiated, proliferative state,
capable of self-renewal, through the suppression of DNMT1
expression during long-term subculture in vitro.
Importantly, the use of HuAECs, in contrast to MEFs,
as feeder layers avoids contamination from heterogeneous proteins.
To date, numerous studies have indicated that the in vivo
proliferation and differentiation of human iPS cells is dependent
on a specific microenvironment, including various cytokines, LIF
and other unknown factors (3,21,37).
In order to maintain the self-renewal and proliferative properties
and to inhibit the differentiation of iPS cells in vitro, a
similar micro-environment must be provided with the essential
ingredients for growth. HuAECs are temporary specialized fetal
cells, derived from the placenta, which are able to maintain the
pluripotency of early epiblast cells. Previous studies have
indicated that HuAECs express a number of growth factors, such as
LIF, EGF, bFGF, transforming growth factor (TGF)-α/β and bone
morphogenetic protein (BMP)-4, as well as stem cell markers,
including Nanog, Oct4 and nestin (20,44).
The results of our previous studies suggested that the expression
of LIF by HuAECs was able to maintain mouse and human ESCs and
mouse spermatogonial stem cells in an undifferentiated,
proliferative state, capable of self-renewal (19,39,40).
Additional advantages of the human placental amnion include its low
toxicity and high safety, due to the presence of few exogenous
foreign proteins. Furthermore, unlike ESCs, its use is free from
ethical constraints. Therefore, this present study demonstrated
that the human placental amnion may be used as an abundant source
of feeder cells for human iPS cultures.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (no.
81202811) and Shanghai Municipal Health Bureau Fund (no. 20124320)
to Dr Te Liu.
References
|
1.
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Takahashi K, Tanabe K, Ohnuki M, et al:
Induction of pluripotent stem cells from adult human fibroblasts by
defined factors. Cell. 131:861–872. 2007. View Article : Google Scholar
|
|
3.
|
Galach M and Utikal J: From skin to the
treatment of diseases - the possibilities of iPS cell research in
dermatology. Exp Dermatol. 20:523–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Maherali N, Sridharan R, Xie W, et al:
Directly reprogrammed fibroblasts show global epigenetic remodeling
and widespread tissue contribution. Cell Stem Cell. 1:55–70.
2007.PubMed/NCBI
|
|
5.
|
Okita K, Ichisaka T and Yamanaka S:
Generation of germline-competent induced pluripotent stem cells.
Nature. 448:313–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hanna J, Markoulaki S, Schorderet P, et
al: Direct reprogramming of terminally differentiated mature B
lymphocytes to pluripotency. Cell. 133:250–264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wernig M, Meissner A, Foreman R, et al: In
vitro reprogramming of fibroblasts into a pluripotent ES-cell-like
state. Nature. 448:318–324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Liu H, Zhu F, Yong J, et al: Generation of
induced pluripotent stem cells from adult rhesus monkey
fibroblasts. Cell Stem Cell. 3:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yu J, Vodyanik MA, Smuga-Otto K, et al:
Induced pluripotent stem cell lines derived from human somatic
cells. Science. 318:1917–1920. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Thier M, Munst B and Edenhofer F:
Exploring refined conditions for reprogramming cells by recombinant
Oct4 protein. Int J Dev Biol. 54:1713–1721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhong B, Watts KL, Gori JL, et al:
Safeguarding nonhuman primate iPS cells with suicide genes. Mol
Ther. 19:1667–1675. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ye Z, Zhan H, Mali P, et al: Human-induced
pluripotent stem cells from blood cells of healthy donors and
patients with acquired blood disorders. Blood. 114:5473–5480. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ezashi T, Telugu BP, Alexenko AP, Sachdev
S, Sinha S and Roberts RM: Derivation of induced pluripotent stem
cells from pig somatic cells. Proc Natl Acad Sci USA.
106:10993–10998. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gonzalez F, Barragan Monasterio M,
Tiscornia G, et al: Generation of mouse-induced pluripotent stem
cells by transient expression of a single nonviral polycistronic
vector. Proc Natl Acad Sci USA. 106:8918–8922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kaji K, Norrby K, Paca A, Mileikovsky M,
Mohseni P and Woltjen K: Virus-free induction of pluripotency and
subsequent excision of reprogramming factors. Nature. 458:771–775.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Zhou H, Wu S, Joo JY, et al: Generation of
induced pluripotent stem cells using recombinant proteins. Cell
Stem Cell. 4:381–384. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yakubov E, Rechavi G, Rozenblatt S and
Givol D: Reprogramming of human fibroblasts to pluripotent stem
cells using mRNA of four transcription factors. Biochem Biophys Res
Commun. 394:189–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Liu T, Cheng W, Liu T, et al: Human
amniotic epithelial cell feeder layers maintain mouse embryonic
stem cell pluripotency via epigenetic regulation of the c-Myc
promoter. Acta Biochim Biophys Sin (Shanghai). 42:109–115. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Liu T, Guo L, Liu Z and Cheng W: Human
amniotic epithelial cells maintain mouse spermatogonial stem cells
in an undifferentiated state due to high leukemia inhibitor factor
(LIF) expression. In Vitro Cell Dev Biol Anim. 47:318–326. 2011.
View Article : Google Scholar
|
|
20.
|
Liu T, Wu J, Huang Q, et al: Human
amniotic epithelial cells ameliorate behavioral dysfunction and
reduce infarct size in the rat middle cerebral artery occlusion
model. Shock. 29:603–611. 2008.
|
|
21.
|
Liu T, Cheng W, Huang Y, Huang Q, Jiang L
and Guo L: Human amniotic epithelial cell feeder layers maintain
human iPS cell pluripotency via inhibited endogenous microRNA-145
and increased Sox2 expression. Exp Cell Res. 318:424–434. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hattori N, Imao Y, Nishino K, et al:
Epigenetic regulation of Nanog gene in embryonic stem and
trophoblast stem cells. Genes Cells. 12:387–396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Cowan CA, Atienza J, Melton DA and Eggan
K: Nuclear reprogramming of somatic cells after fusion with human
embryonic stem cells. Science. 309:1369–1373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Freberg CT, Dahl JA, Timoskainen S and
Collas P: Epigenetic reprogramming of OCT4 and NANOG regulatory
regions by embryonal carcinoma cell extract. Mol Biol Cell.
18:1543–1553. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Simonsson S and Gurdon J: DNA
demethylation is necessary for the epigenetic reprogramming of
somatic cell nuclei. Nat Cell Biol. 6:984–990. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Tada M, Tada T, Lefebvre L, Barton SC and
Surani MA: Embryonic germ cells induce epigenetic reprogramming of
somatic nucleus in hybrid cells. EMBO J. 16:6510–6520. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Bibikova M, Chudin E, Wu B, et al: Human
embryonic stem cells have a unique epigenetic signature. Genome
Res. 16:1075–1083. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Li JY, Pu MT, Hirasawa R, et al:
Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in
the methylation of Oct4 and Nanog. Mol Cell Biol. 27:8748–8759.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Delgado-Olguin P and Recillas-Targa F:
Chromatin structure of pluripotent stem cells and induced
pluripotent stem cells. Brief Funct Genomics. 10:37–49. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Athanasiadou R, de Sousa D, Myant K,
Merusi C, Stancheva I and Bird A: Targeting of de novo DNA
methylation throughout the Oct-4 gene regulatory region in
differentiating embryonic stem cells. PLoS One. 5:e99372010.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tsumura A, Hayakawa T, Kumaki Y, et al:
Maintenance of self-renewal ability of mouse embryonic stem cells
in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b.
Genes Cells. 11:805–814. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Fatemi M, Hermann A, Pradhan S and Jeltsch
A: The activity of the murine DNA methyltransferase Dnmt1 is
controlled by interaction of the catalytic domain with the
N-terminal part of the enzyme leading to an allosteric activation
of the enzyme after binding to methylated DNA. J Mol Biol.
309:1189–1199. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Sen GL, Reuter JA, Webster DE, Zhu L and
Khavari PA: DNMT1 maintains progenitor function in self-renewing
somatic tissue. Nature. 463:563–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Mikkelsen TS, Hanna J, Zhang X, et al:
Dissecting direct reprogramming through integrative genomic
analysis. Nature. 454:49–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Datta J, Ghoshal K, Sharma SM, Tajima S
and Jacob ST: Biochemical fractionation reveals association of DNA
methyltransferase (Dnmt) 3b with Dnmt1 and that of Dnmt 3a with a
histone H3 methyltransferase and Hdac1. J Cell Biochem. 88:855–864.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ohi Y, Qin H, Hong C, et al: Incomplete
DNA methylation underlies a transcriptional memory of somatic cells
in human iPS cells. Nat Cell Biol. 13:541–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Liu T, Zou G, Gao Y, et al: High
efficiency of reprogramming CD34+ cells derived from
human amniotic fluid into induced pluripotent stem cells with Oct4.
Stem Cells Dev. 21:2322–2332. 2012.PubMed/NCBI
|
|
38.
|
Miyabayashi T, Teo JL, Yamamoto M,
McMillan M, Nguyen C and Kahn M: Wnt/beta-catenin/CBP signaling
maintains long-term murine embryonic stem cell pluripotency. Proc
Natl Acad Sci USA. 104:5668–5673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Lai D, Cheng W and Liu T, Jiang L, Huang Q
and Liu T: Use of human amnion epithelial cells as a feeder layer
to support undifferentiated growth of mouse embryonic stem cells.
Cloning Stem Cells. 11:331–340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Lai D, Cheng W, Liu T, Jiang L, Liu T,
Huang Q and Guo L: Optimization of culture conditions to support
undifferentiated growth of human embryonic stem cells. Cell
Reprogram. 12:305–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Guo Y, Liu S, Wang P, et al: Expression
profile of embryonic stem cell-associated genes Oct4, Sox2 and
Nanog in human gliomas. Histopathology. 59:763–775. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Robertson KD, Uzvolgyi E, Liang G, et al:
The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate
mRNA expression in normal tissues and overexpression in tumors.
Nucleic Acids Res. 27:2291–2298. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Liu T, Chen Q, Huang Y, Huang Q, Jiang L
and Guo L: Low microRNA-199a expression in human amniotic
epithelial cell feeder layers maintains human-induced pluripotent
stem cell pluripotency via increased leukemia inhibitory factor
expression. Acta Biochim Biophys Sin (Shanghai). 44:197–206. 2012.
View Article : Google Scholar
|
|
44.
|
Miki T, Lehmann T, Cai H, Stolz DB and
Strom SC: Stem cell characteristics of amniotic epithelial cells.
Stem Cells. 23:1549–1559. 2005. View Article : Google Scholar : PubMed/NCBI
|