Introduction
Primary renal glucosuria (PRG; OMIM #233100;
http://omim.org/entry/233100. Accessed
August 5, 2012) is characterized by persistent glucosuria due to a
reduction in the renal tubular reabsorption of glucose in the
presence of a normal concentration of serum glucose and the absence
of any other impairment of tubular function. It is caused by
mutations in the SLC5A2 gene on chromosome 16p11.2, which
codes for the low-affinity high-capacity sodium/glucose
co-transporter SGLT2 (1). This
transporter is responsible for the tubular reabsorption of filtered
glucose. Numerous case reports have confirmed that SLC5A2
mutations are responsible for the majority of cases of PRG
(2–7).
To date, >50 different mutations of the
SLC5A2 gene have been reported, the majority of which are
restricted to a single individual or family. Although PRG is known
to be inherited as an autosomal recessive trait, certain cases of
PRG have demonstrated co-dominance or a pattern of autosomal
dominance. Numerous heterozygous individuals have mild glucosuria,
whereas homozygous or compound heterozygous mutations normally have
massive glucosuria >10 g/1.73 m2/24 h. Co-dominance
with incomplete penetrance supports the best fit for the
inheritance pattern of PRG (2).
In the current study, the clinical and genetic
findings of a subject with PRG associated with a novel mutation of
the SLC5A2 gene are described. The well-being of the patient
suggests that PRG may have a benign nature.
Case report
The subject of this study is a healthy 40-year-old
man, the fifth child of Korean parents. The parents of the patient
have no history of glucosuria. The patient had exhibited glycosuria
in the absence of hyperglycemia for >20 years and attended
Soonchunhyang University Bucheon Hospital (Bucheon, Korea) for an
evaluation. The patient was noted to have glucosuria at a routine
urinalysis check-up. The patient had no history of diabetes or
hypertension, or of cardiac, pulmonary, hepatic, renal or
musculoskeletal disorders, and exhibited no evidence of
hypoglycemia. Furthermore, the patient exhibited no urological
manifestations related to renal glucosuria. Work-ups repeated in
order to elucidate the cause of the persistent glucosuria failed.
The patient reported that two older brothers had a history of
glucosuria.
An extensive laboratory work-up was performed in
order to investigate the cause of the patient’s renal glucosuria.
Urine amino acid analysis, 24-h urine chemistry and urine
osmolality were performed.
Molecular defects in the SLC5A2 gene were
investigated to confirm the diagnosis of PRG. After obtaining
informed consent from the patient, blood samples were collected.
Genomic DNA was isolated from peripheral blood leukocytes using a
Wizard genomic DNA purification kit according to the manufacturer’s
instructions (Promega, Madison, WI, USA). The SLC5A2 gene
was amplified by polymerase chain reaction (PCR) using primers
designed by the authors (forward: 5′-ACAACGGTCTAAGGCGCAGTC-3′,
reverse: 5′-TTAGGAGGGTGACGGAACTGG-3′) and a Thermal Cycler 9700
(Applied Biosystems, Foster City, CA, USA). Sequence analysis of
all coding exons and the flanking introns of the SLC5A2 gene
were performed using the BigDye Terminator Cycle Sequencing Ready
Reaction kit (Applied Biosystems) on an ABI Prism 3130 genetic
analyzer (Applied Biosystems). Nucleotide numbering reflects cDNA
numbering with c.1 corresponding to A of the ATG translation
initiation codon in the reference sequence of SLC5A2 (NM_003041.3).
Potential mutations were defined by their exclusion from the Human
Gene Mutation Database (http://www.hgmd.cf.ac.uk) and previously reported
mutations on PubMed (http://ncbi.nlm.nih.gov/PubMed/). All novel mutations
were confirmed by sequencing 100 control chromosomes. The study was
approved by the ethics committee of the Institutional Review Board
and also the screening of the SLC5A2 gene in normal subjects was
approved by the ethics committee of Soonchunhyang University
Bucheon Hospital (Bucheon, Korea).
Results
The patient exhibited a fasting blood sugar level of
104 mg/dl [reference range (RR), 60–108 mg/dl], a 2-h postprandial
sugar level of 101 mg/dl, a sodium level of 144 mmol/l (RR, 135–145
mmol/l), a potassium level of 3.7 mmol/l (RR, 3.5–5.5 mmol/l), and
a chloride level of 106 mmol/l (RR, 98–110 mmol/l) in serum.
Twenty-four-hour urine chemistry revealed that the quantity of
glucose excreted was 10.8 g/1.73 m2/24 h (RR, <0.5
g/1.73 m2/24 h). However, protein, uric acid, urea
nitrogen, creatinine, sodium, potassium, chloride, calcium and
phosphorus levels were within the RR. The osmolality of urine was
763 mOsm/kg (RR, 300–900 mOsm/kg), and amino acid analysis in urine
revealed no amino aciduria. There was no evidence of proteinuria,
acidosis, β-2 microglobulinuria, acidosis or phosphaturia.
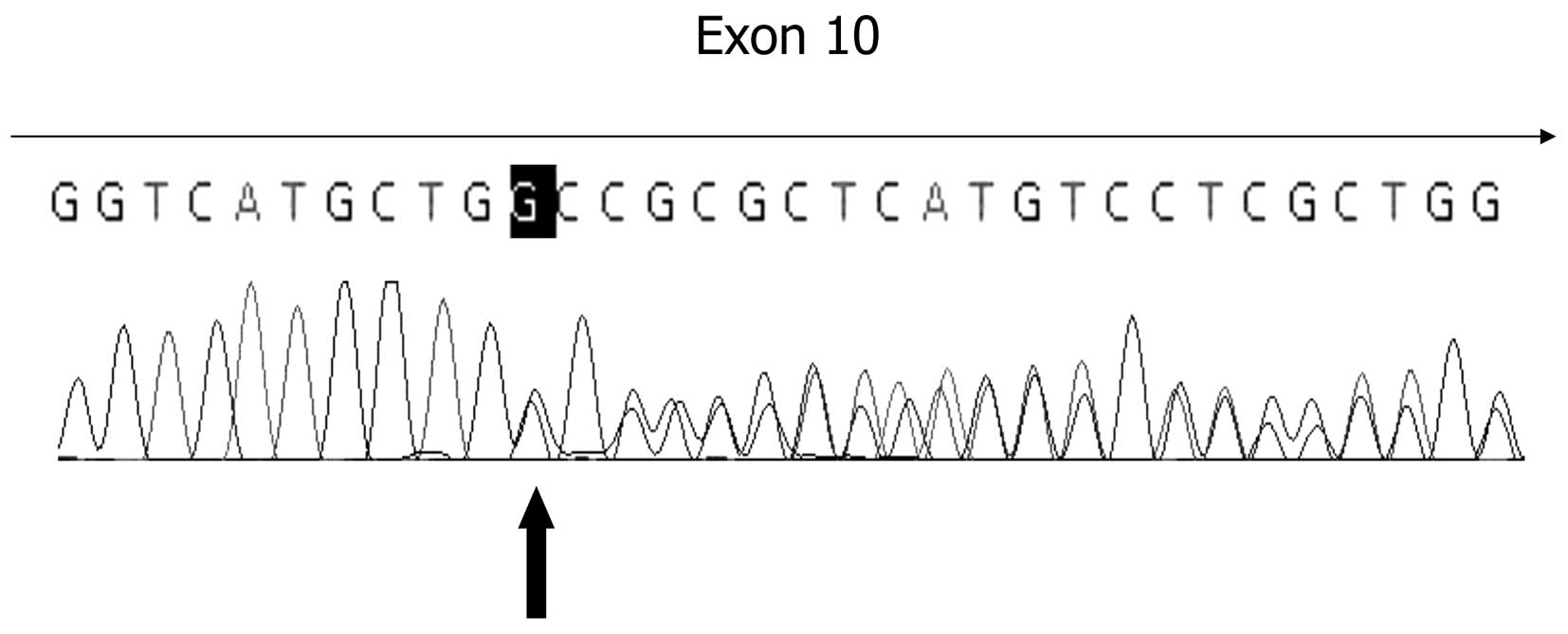
Direct sequencing of the SLC5A2 gene from the
patient with PRG revealed a novel 1 bp deletion mutation, altering
the coding sequence of exon 10 in the transmembrane domain
(c.1162delG; Ala388ProfsX48; Fig.
1). The 1 bp deletion results in the frameshift mutation
starting from 388th codon, which is truncated at the 48th codon
from Ala388. Screening of the SLC5A2 gene in 50 normal
control subjects revealed no mutant alleles in 100 screened
chromosomes. SLC5A2 gene analysis suggested an autosomal
dominant inheritance pattern.
Discussion
In this study, a novel deletion mutation in the
SLC5A2 gene in a patient with PRG, associated with benign
clinical characteristics, is reported. The patient appeared healthy
despite having had glucosuria since childhood and exhibited no ill
effects of the glucosuria.
We speculate that glucosuria caused by carrying the
SLC5A2 gene mutation may have a potentially beneficial
effect on the healthy state. The persistent loss of calories and
sodium may have a beneficial effect on lowering blood pressure and
preventing obesity, although the majority of the sodium is
reabsorbed by distal tubules (8).
A pharmacological inhibitor of SGLT2 (sodium-glucose
co-transporters inhibitor) has been evaluated as a useful drug for
the treatment of obesity, diabetes, and hypertension with no
significant toxic effects (9).
Among sodium/glucose transporters, SGLT2 is
expressed almost exclusively in the kidney (10) and is encoded by the SLC5A2
gene, which is responsible for PRG. A large number of heterogeneous
mutations have been reported in the SLC5A2 gene. However,
previous reports have not observed an identifiable mutation in the
SLC5A2 gene (2,3). This may be due to technical
limitations related to mutational analysis. According to the
previous reports, no clear association has been identified between
genotype and phenotype in PRG, with the exception of a tendency for
increased homozygous or compound heterozygous mutation frequency in
patients exhibiting severe glucosuria.
Although PRG is a rare disease, many subjects with
glucosuria without hyperglycemia have been confirmed by
SLC5A2 gene analysis, as in the current case. In the present
case, the c.1162delG mutation affects exon 10, including an alanine
residue conserved in the entire transporter superfamily to which
SGLT2 belongs, abolishing the expression of the glucose
transporter. This finding is in agreement with previous studies,
suggesting that SLC5A2 mutations have marked allelic
heterogeneity, and the majority of mutations in PRG are restricted
to a single individual or family (11).
Unfortunately, it was not possible to perform a
family study; therefore, an evaluation of the mutational pattern of
the patient to determine whether the mutation was de novo or
inherited could not be conducted. Furthermore, it was not possible
to induce information regarding the genotype-phenotype correlation,
due to there only being one case. Despite the large deletion
involving the SLC5A2 gene, the majority of mutations may be
detected by sequencing analysis. As previously reported, compound
heterozygotes that carry missense or truncated mutations
consistently exhibited renal glucosuria due to SGLT2 dysfunction
(2–7). Therefore, identification of the
mutation that causes glucosuria may enable the establishment of a
genotypic diagnosis of PRG, providing important information to
families and physicians.
In conclusion, PRG should be considered when making
a differential diagnosis of patients exhibiting unexplained renal
glucosuria without hyperglycemia or hypoglycemia, and SLC5A2
gene analysis should be performed. Molecular diagnosis may be
beneficial for the confirmation of renal glucosuria and in deciding
interventional measures for the patient’s family members.
Acknowledgements
This study was supported by the Soonchunhyang
University Research Fund.
References
|
1
|
van den Heuvel LP, Assink K, Willemsen M
and Monnens L: Autosomal recessive renal glucosuria attributable to
a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet.
111:544–547. 2002.PubMed/NCBI
|
|
2
|
Santer R, Kinner M, Lassen CL, et al:
Molecular analysis of the SGLT2 gene in patients with renal
glucosuria. J Am Soc Nephrol. 14:2873–2882. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calado J, Loeffler J, Sakallioglu O, et
al: Familial renal glucosuria: SLC5A2 mutation analysis and
evidence of salt-wasting. Kidney Int. 69:852–855. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calado J, Soto K, Clemente C, Correia P
and Rueff J: Novel compound heterozygous mutations in SLC5A2 are
responsible for autosomal recessive renal glucosuria. Hum Genet.
114:314–316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kleta R, Stuart C, Gill FA and Gahl WA:
Renal glucosuria due to SGLT2 mutations. Mol Genet Metab. 82:56–58.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Francis J, Zhang J, Farhi A, Carey H and
Geller DS: A novel SGLT2 mutation in a patient with autosomal
recessive renal glucosuria. Nephrol Dial Transplant. 19:2893–2895.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Magen D, Sprecher E, Zelikovic I and
Skorecki K: A novel missense mutation in SLC5A2 encoding SGLT2
underlies autosomal-recessive renal glucosuria and aminoaciduria.
Kidney Int. 67:34–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cruz DN, Simon DB, Nelson-Williams C, et
al: Mutations in the Na-Cl cotransporter reduce blood pressure in
humans. Hypertension. 37:1458–1464. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chao EC and Henry RR: SGLT2 inhibition - a
novel strategy for diabetes treatment. Nat Rev Drug Discov.
9:551–559. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wells RG, Pajor AM, Kanai Y, Turk E,
Wright EM and Hediger MA: Cloning of a human kidney cDNA with
similarity to the sodium-glucose cotransporter. Am J Physiol.
263:F459–465. 1992.PubMed/NCBI
|
|
11
|
Calado J, Sznajer Y, Metzger D, et al:
Twenty-one additional cases of familial renal glucosuria: absence
of genetic heterogeneity, high prevalence of private mutations and
further evidence of volume depletion. Nephrol Dial Transplant.
23:3874–3879. 2008.PubMed/NCBI
|