Introduction
It is well known that atherosclerosis (AS),
characterized by an infiltration of leukocytes into the lesion
foci, is one of the most widespread threats to human health and
survival (1). The formation and
development of AS lesions is a chronic and progressive process that
requires a long time. AS is characterized by the accumulation of
lipids and other elements in the coronary artery (2–5).
However, the precise etiopathogenesis of the disease is currently
unknown. The mechanisms behind the development of AS may include
dyslipidemia and inflammation among other factors.
Fibrinogen (Fg) plays a significant role in
homeostasis and thrombosis and is able to promote the formation of
atherosclerotic plaques through various mechanisms. Fg promotes
cell migration and adhesion. In addition, large amounts of fibrin,
a metabolite of Fg, are located in atherosclerotic plaques and also
promote the proliferation and migration of cells. Fibrin is also
able to bind to fibronectin. Finally, fibrin in the inner layer is
able to attract leukocytes and promote lipid accumulation in
atherosclerotic plaques (6).
Platelets and P-selectin also play significant roles in hemostasis
and thrombosis. Evidence has shown that P-selectin is able to
promote the development of atherosclerotic lesions (1,3,7). A
study by Yang et al(7)
reported for the first time that Fg may restore the surface
expression of P-selectin in Fg-deficient (Fg-/-) mice. We thus
hypothesized that Fg may regulate or control the expression of
P-selectin during the formation and/or development of AS and thus
facilitate atherosclerotic lesion development and promote plaque
formation. In the present study, AS was successfully induced in
Sprague-Dawley (SD) rats prior to Fg being trans-fused into them.
Fg was shown to promote the development of atherosclerotic lesions
and plaques, as rats that did not receive an Fg transfusion
developed atherosclerotic lesions that were relatively small in
comparison.
Materials and methods
Animal breeding and experimental
protocol
SD rats (SPF, 200±10 g) were purchased from the
Experimental Animal Center of Anhui, China. The rats were
maintained in controlled temperature (21–23°C), light (12-h light,
12-h dark) and humidity (55±5%) conditions with access to food and
water ad libitum. Subsequent to a 3-day adaptation period,
they were randomly divided into 4 groups. Groups Z and ZF were fed
a normal chow diet, while groups H and HF were fed a high-fat diet
for ∼15 weeks. The high-fat diet contained 83.3% normal chow, 8%
lard, 3% cholesterol, 5% plantation white sugar, 0.2%
propylthiouracil and 0.5% chleolate. At the beginning of the
experiment, vitamin D2 (3×105 U/kg) was
injected into the rats from group H and HF. In the 7th and 8th
weeks, human Fg was injected intravenously at a dose of 2.0 mg per
rat into the rats from groups ZF and HF. At the end of the 15th
week, all animals were fasted for ≥8 h prior to being anesthetized
with 10% chloral hydrate at a dose of 0.3 ml/100 g. Blood was
obtained from the common abdominal aorta of the rats and
biochemical characteristics were then measured and enzyme-linked
immunosorbent assays (ELISAs) performed. The aortas were carefully
separated, removed, cut open, observed and placed in 10% (w/v)
neutral formalin for later use. All the animal experiments were
conducted with approval from the Internal Animal Care and Use
Committee of Anhui Medical University and in compliance with the
Guide for the Care and Use of Laboratory Animals.
Reagents
Unconjugated anti-rat P-selectin antibodies were
purchased from Boiss (Beijing, China). Unconjugated anti-rat
fibrinogen antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). SP-9000/9001/9002 Histostain TM-Plus
kits were purchased from Zymed (San Diego, CA, USA). All other
chemicals used were commercially available and pure grade.
Biochemical analyses
The blood fat concentrations were determined
enzymatically using automatic biochemical analyzers.
Morphological measurements (HE
staining)
The aortas were stored in 10% (w/v) neutral formalin
for at least one day as described. The roots of the aortas were cut
off and then the samples were washed, dehydrated, cleared, dipped
in wax, embedded, sliced, coated, grilled and stained with HE.
Finally, the thickness of the intima and media were examined and
photographed with an image operation system under a 1/10-mm eye
lens and with 40×10 amplification.
Immunohistochemistry
The aorta slices were dewaxed and washed with
distilled water and then incubated in 3% hydrogen peroxide for 10
min to block the endogenous peroxidase. Subsequent to being washed
with PBS, the aorta slices were incubated in 10% normal goat serum
for 30 min to block unspecific binding. Mouse monoclonal primary
antibodies against P-selectin or Fg were added and incubated
overnight at 4°C. The samples were placed at 37°C for 30 min to
return them to a normal temperature and then were incubated with a
secondary antibody for 20 min at 37°C, followed by being washed 3
times with PBS for 3 min each time. Horseradish peroxidase (HRP;
100 μl) was added to the slides which were then incubated at
37°C for 20 min. Following coloration with diaminobenzidine (DAB),
the slides were processed with hematoxylin light staining for 2
min, followed by bluing, dehydration, clearing and mounting.
Finally, the morphological changes in the vessel walls were
observed and images were captured.
Detection of plasma Fg by ELISA
Plasma Fg was measured using an Fg ELISA kit
(R&D Systems, Minneapolis, MN, USA). The blood samples were
collected into EDTA-coated cuvettes and centrifuged at 1,000 x g
for 10 min to remove the cells and collect the plasma. All reagents
were prepared prior to starting the assay procedure. As recommended
by the manufacturer, all standards and samples were added in
duplicate to the microELISA strip plate. A total of 50 μl of
each standard and 10 μl of the testing samples diluted in 40
μl of the sample dilution were used. A blank well with
nothing added was also included. Next, 100 μl of the
HRP-conjugate reagent was added to each well and the plates were
covered with an adhesive strip and incubated for 60 min at 37°C.
Each well was aspirated and washed five times by filling the well
with a wash solution (400 μl) using a squirt bottle,
manifold dispenser or autowasher. The liquid was removed completely
at each step to ensure a good performance. Subsequent to the last
wash, any remaining wash solution was removed by aspirating or
decanting. The plate was then inverted and blotted against clean
paper towels. Next, 50 μl chromogen solution A and 50
μl chromogen solution B were added to each well and the
plate was gently mixed and incubated for 15 min at 37°C in the
dark. After this, 50 μl stop solution was added to each
well. The color in the wells was observed to change from blue to
yellow. If the color in the well was green or the color change did
not appear uniform, the plates were gently tapped to ensure
thorough mixing. The optical density (OD) was read at 450 nm using
a microtiter plate reader within 15 min of the addition.
Statistical analysis
All statistical analyses were performed using SPSS
version 13.0 for Windows (SPSS, Inc., Chicago, IL, USA). All data
are expressed as mean ± standard deviation (SD). Comparisons
between the groups were carried out using a one-way analysis of
variance (ANOVA) and the Student-Newman-Keuls (SNK) method. A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
Changes in the blood fat levels in rats
from the various experimental groups
The induction of hypercholesterolemia was
accompanied by an increase in the serum total cholesterol (TCH) and
low-density lipoprotein cholesterol (LDL-C) levels. The serum
lipoprotein analysis showed a dominant LDL-C fraction. The changes
in the cholesterol and glucose levels of the rats following the
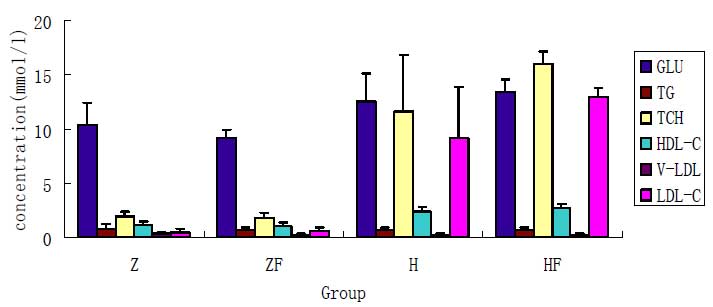
various treatments for each group are shown in Fig. 1. As shown in Fig. 1, the serum TCH and LDL-C levels in
the group Z and ZF were significantly lower than those of the group
H and HF (P<0.05), there was statistical significance
(P<0.05). However, there was no statistically significant
difference in the serum levels of TCH and LDL-C between groups H
and HF or groups Z and ZF.
Plasma Fg levels
The plasma fibrinogen level was detected by ELISA in
the four groups, there was significant differences in group H or
group HF compared with group Z, there was a significant difference
between group HF and group ZF, there was no difference between
groups ZF and Z or groups HF and H (Table I).
 | Table I.Plasma Fg (g/l) levels in rats from
the various experimental groups (mean ± SD). |
Table I.
Plasma Fg (g/l) levels in rats from
the various experimental groups (mean ± SD).
| Group | Fg |
|---|
| Z | 2.25±0.25 |
| ZF | 3.09±0.20 |
| H | 3.72±0.23a |
| HF |
4.21±0.35a,b |
Light microscopic analysis of the
pathological HE-stained sections
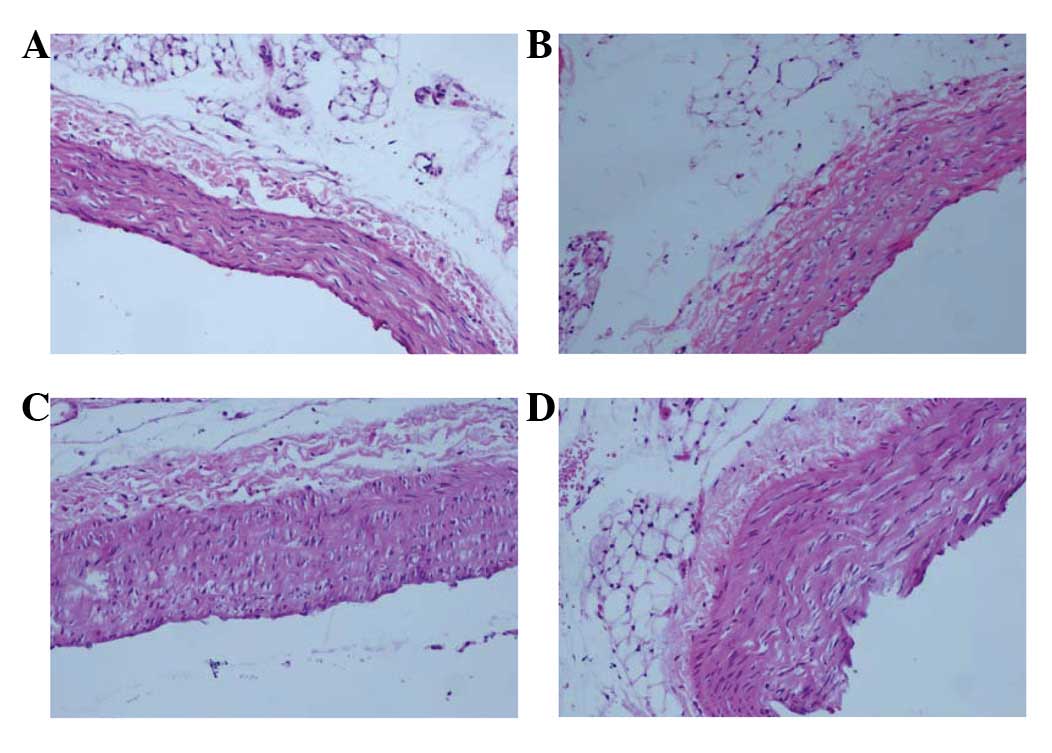
In the control group, group Z, the vessel walls were
round and even in thickness. The inner and outer elastic plates
were clear and complete and the endotheliocyte core was stained
blue and evenly arranged. Also, no smooth muscle cells were
observed underneath the endoderm (Fig.
2A). The vessel walls in group ZF were not as smooth as in
group Z, but no foam cells were observed (Fig. 2B). The vessels in group H were
rougher and thicker than those in groups Z and ZF and numerous foam
and inflammatory cells were detected (Fig. 2C). The vessel walls in group HF
were rough, the intima exhibited signs of hyperplasia and the
thickness was uneven compared with group H. Numerous foam cells and
atheronecrotic substances were observed under the fiber caps and
cholesterol crystals and a few inflammatory cells were also
observed there (Fig. 2D).
Immunohistochemical staining of Fg and
P-selectin
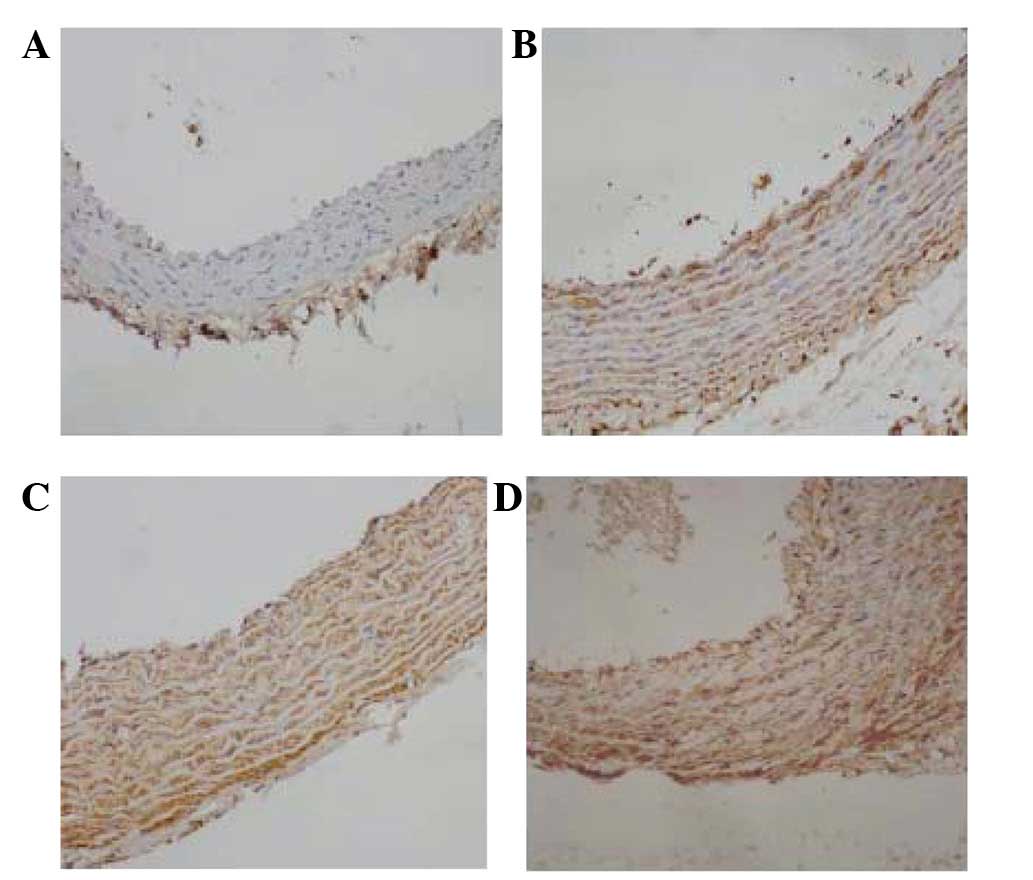
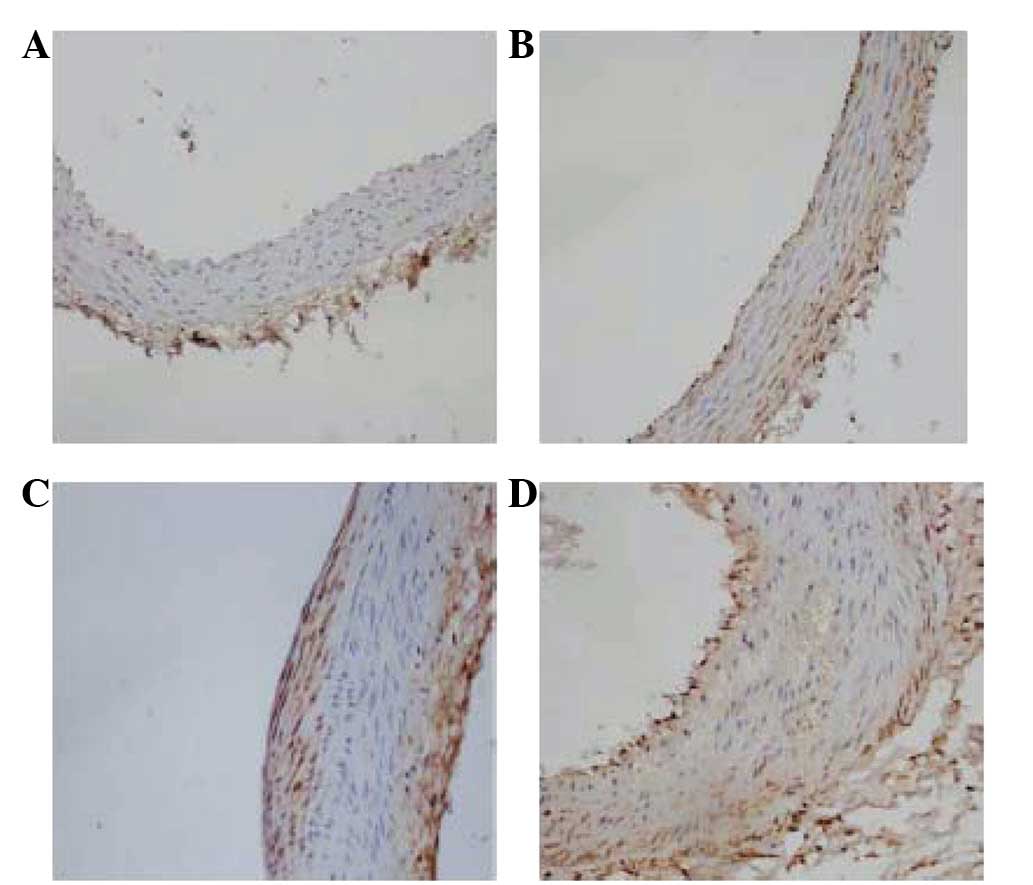
Immunohistochemistry was performed for Fg or
P-selectin on representative tissue sections. Positive Fg or
P-selectin staining was observed as a brown stain. In group Z,
little brown staining was observed in the vessel walls,
particularly in the endarterium, while a small amount of brown
staining was observed underneath the endoderm in the rats of the ZF
group (Figs. 3A and B and 4A and B). In group H, lots of positive
staining was detected in the vessel walls (Figs. 3C and 4C). In group HF, Fg or P-selectin
immunostaining resulted in a more widespread and dense smear,
including brown staining in the nucleus (Figs. 3D and 4D).
Discussion
Currently, cardiovascular and cerebrovascular
disease, of which AS is a component, are two major causes of
disability and mortality (2,3). The
investigation into the etiopathogenesis and pathogenesis of AS and
the development of effective measures to delay and reverse the
progression of AS, thereby reducing mortality from cardiovascular
and cerebrovascular diseases, has become an important field of
study (8–10). However, AS has a complex,
multifactorial pathophysiology and a number of risk factors work
together to shape atherosclerotic plaques. The initial stage of AS
is characterized by the infiltration and adherence of monocytes to
the surface of the injured endothelium, for which adhesion
molecules maybe indispensable.
Fg, which is produced by hepatocytes, is composed of
three homologous polypeptide chains; α, β and γ. As the soluble
precursor of fibrin, Fg is a 340-kDa hexamer (11) that has a central E domain, two
peripheral D domains and three stranded coiled coils. The D domain
contains a globular carboxyl end made up of β- and γ-chains and the
E domain contains an α-terminus with 6 polypeptide chains. The E
and D domains are linked by three stranded coiled coils (12,13).
Fg is a key molecule in hemostasis and thrombosis and also plays a
role in pathophysiological processes, including infection and wound
healing. Fg also plays a significant role in the formation and
progression of atherosclerotic plaques (12,14–18).
It is becoming increasingly clear that fibrinogen is an
inflammation marker for cardiovascular disease (6) that is expressed at the site of plaque
ruptures (16).
It is also evident that Fg plays a significant role
in AS. Certain possible mechanisms by which Fg affects AS have been
identified (17,19). Firstly, Fg-fibrin composition is
able to promote the formation of atherosclerotic plaques in AS by
enhancing the deposition of lipids into the vessel walls, thereby
attracting macrophagocytes which swallow the lipid material
(12) and form foam cells.
Secondly, Fg plays a key role in thrombosis, which causes plaque
instability in AS. Thirdly, Fg is able to contribute to
atherogenesis through interactions with the endothelial cells,
smooth muscle cells and macrophages, while also playing a role in
the transfer of cholesterol to the mononuclear cells and
macrophages (19,20). Fg and fibrin are able to stimulate
chemokine secretion and facilitate neutrophil-endothelial cell
interactions. Numerous clinical experiments have demonstrated that
Fg plays a critical role in the formation of plaques. Lepedda et
al(16) reported that patients
with unstable plaques had a higher level of plasma Fg than patients
with stable plaques. This means that Fg may play a more significant
role in the formation of unstable plaques than in stable plaques. A
previous study demonstrated that an infusion of activated platelets
caused the release of Weibel-Palade bodies leading to
P-selectin-mediated leukocyte rolling. This suggested that
P-selectin is crucial in the processes of inflammation and AS
(15–17). Earlier studies (21,22,16,18)
have clearly demonstrated that Fg enhances intracellular platelet
P-selectin levels and affects P-selectin expression on the surface
of mouse and human platelets. This may partially explain the role
of Fg in inflammation, hemostasis and AS. P-selectin is a member of
the selectin family of cell adhesion receptors and is also known as
CD62P, GMP-140 or PADGEM (platelet activation-dependent granule
external membrane) (23).
P-selectin is localized to Weibel-Palade bodies in the endothelial
cells or to α-granules in platelets (24). Platelet P-selectin is involved in
multiple physiological processes, including platelet aggregation
and platelet-leukocyte and platelet-endothelial cell interactions.
Clinically, P-selectin is widely accepted as a marker of platelet
activation, with the elevated levels of plasma P-selectin in
thrombotic disorders resulting mainly from the shedding of
P-selectin from the surface of the platelets. Studies have
confirmed that P-selectin gene-deficient mice have a lower
incidence of AS (25,26). Graff et al(27) discovered that P-selectin levels are
highly associated with the release of platelet-derived growth
factor (PDGF), which is one of the growth factors secreted by
endothelial cells (ECs). The authors also confirmed that P-selectin
may participate in the early stages of AS. However, there is as of
yet little insight into the mechanisms that regulate platelet
P-selectin expression. In the present study, Fg transfusion in the
experimental ZF group led to a higher level of P-selectin
expression compared with that observed in group Z, as determined by
immunohistochemistry. Similar results were also obtained when
groups H and HF were compared.
P-selectin may contribute to the formation of AS in
several ways. P-selectin expression is associated with the adhesion
of mononuclear cells to vessel walls and the subsequent formation
of fatty streaks. P-selectin-mediated macrophage and T cell
accumulation in the endarterium and platelet activation are also
involved in the complications associated with AS. Variations in the
sheer stress increase platelet P-selectin levels. The deposition of
platelets into the extracellular matrix provides a leading adhesion
site for the accumulation of molecules. The present study observed
that it is easier to form plaques in certain crotch of grave
vessels, this may be associated with the activation and deposition
of platelets that were caused by the changes in hemodynamics and
the high P-selectin expression (28,29).
In the present study, the expression of Fg and
P-selectin in an atherosclerotic SD rat model system was examined.
Differences in the Fg levels may account for the differences
observed in the P-selectin levels, although the possibility of
other effects attributed to Fg cannot be excluded. Fg was
demonstrated as able to promote the development of AS lesions,
while P-selectin levels, which also play a role in lesion
development, were identified as positively correlated with Fg
levels. Thus, we propose that affecting P-selectin expression
levels may be one mechanism by which Fg participates in AS. These
two factors may have a synergistic effect on the development of
lesions and AS. It is therefore worthwhile to further investigate
the correlation between Fg and platelet P-selectin expression in
AS. In addition, variations in Fg concentration may significantly
affect intracellular and cell surface platelet P-selectin
expression.
In summary, Fg and P-selectin are crucial for the
growth of atherosclerotic lesions. Thus, agents that inhibit Fg
and/or P-selectin or that prevent platelet activation may become
effective tools in reducing the development of atherosclerotic
lesions. Moreover, further demonstrations of whether Fg actually
affects P-selectin levels in AS, as well as a clarification of the
mechanism behind such an action, require additional investigation.
Further studies should also be conducted into whether other factors
affect Fg and P-selectin expression or function.
Acknowledgements
This study was supported by a Doctoral
Fund of the Ministry of Education of China (No. 20103420110001),
the Intercollegiate Key Project of Nature Science of Anhui Province
(No. KJ2011A158) and the Youth Research Program of Anhui Provincial
Health Department (No. 09B106). The authors would like to thank Dr
Heyu Ni for his advice during the experiments.
References
|
1.
|
Burger PC and Wagner DD: Platelet
P-selectin facilitates athero-sclerotic lesion development. Blood.
101:2661–2666. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Murray CJ and Lopez AD: Global mortality,
disability, and the contribution of risk factors: Global Burden of
Disease Study. Lancet. 349:1436–1442. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hu MY, Li YL, Jiang CH, Liu ZQ, Qu SL and
Huang YM: Comparison of lycopene and fluvastatin effects on
atherosclerosis induced by a high-fat diet in rabbits. Nutrition.
24:1030–1038. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Papageorgiou N, Tousoulis D, Siasos G and
Stefanadis C: Is fibrinogen a marker of inflammation in coronary
artery disease? Hellenic J Cardiol. 51:1–9. 2010.PubMed/NCBI
|
|
7.
|
Yang H, Lang S, Zhai Z, Li L, Kahr WH,
Chen P, Brkić J, Spring CM, Flick MJ, Degen JL, Freedman J and Ni
H: Fibrinogen is required for maintenance of platelet intracellular
and cell-surface P-selectin expression. Blood. 114:425–436. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Zhai Z, Wu J, Xu X, Ding K, Ni R, Hu W,
Sun Z and Ni H: Fibrinogen controls human platelet fibronectin
internalization and cell-surface retention. J Throm Haemost.
5:1740–1746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Li X and Cong H: Platelet-derived
microparticles and the potential of glycoprotein IIb/IIIa
antagonists in treating acute coronary syndrome. Tex Heart Inst J.
36:134–139. 2009.PubMed/NCBI
|
|
10.
|
Eriksson AC, Jonasson L, Lindahl TL,
Hedbäck B and Whiss PA: Static platelet adhesion, flow cytometry
and serum TXB2 levels for monitoring platelet inhibiting treatment
with ASA and clopidogrel in coronary artery disease: a randomised
cross-over study. J Transl Med. 7:42–56. 2009. View Article : Google Scholar
|
|
11.
|
de Moerloose P and Neerman-Arbez M:
Congenital fibrinogen disorders. Semin Thromb Hemost. 35:356–366.
2009.
|
|
12.
|
de Moerloose P, Boehlen F and
Neerman-Arbez M: Fibrinogen and the risk of thrombosis. Semin
Thromb Hemost. 36:7–17. 2010.
|
|
13.
|
Duga S, Asselta R, Santagostino E, Zeinali
S, Simonic T, Malcorati M, Mannucci PM and Tenchini ML: Missense
mutations in the human beta fibrinogen gene cause congenital
afibrinogenemia by impairing fibrinogen secretion. Blood.
95:1336–1341. 2000.PubMed/NCBI
|
|
14.
|
Green D, Foiles N, Chan C, Schreiner PJ
and Liu K: Elevated fibrinogen levels and subsequent subclinical
atherosclerosis: the CARDIA Study. Atherosclerosis. 202:623–631.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Grebe MT, Luu B, Sedding D, Heidt MC,
Kemkes-Matthes B, Schaefer CA, Tillmanns HH and Gündüz D:
Fibrinogen promotes early atherosclerotic changes of the carotid
artery in young, healthy adults. J Atheroscler Thromb.
17:1003–1008. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lepedda AJ, Cigliano A, Cherchi GM, et al:
A proteomic approach to differentiate histologically classified
stable and unstable plaques from human carotid arteries.
Atherosclerosis. 203:112–118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kannel WB: Overview of hemostatic factors
involved in atherosclerotic cardiovascular disease. Lipids.
40:1215–1220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Koenig W: Fibrin(ogen) in cardiovascular
disease: an update. Thromb Haemost. 89:601–609. 2003.PubMed/NCBI
|
|
19.
|
Retzinger GS, DeAnglis AP and Patuto SJ:
Adsorption of fibrinogen to droplets of liquid hydrophobic phases.
Functionality of the bound protein and biological implications.
Arterioscler Thromb Vasc Biol. 18:1948–1957. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Rabbani LE and Loscalzo J: Recent
observations on the role of hemostatic determinants in the
development of the atherothrombotic plaque. Atherosclerosis.
105:1–7. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
George JN, Lyons RM and Morgan RK:
Membrane changes associated with platelet activation. Exposure of
actin on the platelet surface after thrombin-induced secretion. J
Clin Invest. 66:1–9. 1980. View Article : Google Scholar
|
|
22.
|
Schwertz H, Zimmerman GA and Weyrich AS:
Fibrinogen selects selectins. Blood. 114:2342009. View Article : Google Scholar
|
|
23.
|
Ley K: The role of selectins in
inflammation and disease. Trends Mol Med. 9:263–268. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
McEver RP: Adhesive interactions of
leukocytes, platelets, and the vessel wall during hemostasis and
inflammation. Thromb Haemost. 86:746–756. 2001.PubMed/NCBI
|
|
25.
|
Dong ZM, Brown AA and Wagner DD: Prominent
role of P-selectin in the development of advanced atherosclerosis
in ApoE-deficient mice. Circulation. 101:2290–2295. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Collins RG, Velji R, Guevara NV, Hicks MJ,
Chan L and Beaudet AL: P-Selectin or intercellular adhesion
molecule (ICAM)-1 deficiency substantially protects against
atherosclerosis in apolipoprotein E-deficient mice. J Exp Med.
191:189–194. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Graff J, Klinkhardt U, Schini-Kerth VB,
Harder S, Franz N, Bassus S and Kirchmaier CM: Close relationship
between the platelet activation marker CD62 and the granular
release of platelet-derived growth factor. J Pharmacol Exp Ther.
300:952–957. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Koyama H, Maeno T, Fukumoto S, et al:
Platelet P-selectin expression is associated with atherosclerotic
wall thickness in carotid artery in humans. Circulation.
108:524–529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Libby P, Ridker PM and Hansson GK; Leducq
Transatlantic Network on Atherothrombosis: Inflammation in
atherosclerosis: from pathophysiology to practice. J Am Coll
Cardiol. 54:2129–2138. 2009. View Article : Google Scholar : PubMed/NCBI
|